
7 Epigenetics
Session Level Objectives (SLOs): after completing the session, students will be able to:
SLO1. Understand the fundamentals of chromatin structure and remodeling.
EPIGENETICS
Epigenetics was first defined in the 1940’s by Conrad Waddington as “the branch of biology which studies the causal interactions between genes and their products which bring the phenotype into being.” Today, Epigenetics most commonly refers to heritable changes of gene expression (across cell divisions or across generations) that do not involve changes in DNA sequence.
Epigenetic regulation most commonly occurs as a result of covalent modification of either DNA or of the DNA-packaging histone proteins. We will begin with a discussion of the fundamentals of chromatin structure to provide context for these covalent modifications and their effects. Chromatin is an intrinsically repressive environment for gene expression; much of the DNA is masked by protein. Transcriptional activators can recruit proteins that regulate chromatin accessibility. Cellular mechanisms that control accessibility to DNA in chromatin include covalent DNA modifications, covalent histone modifications, and ATP-dependent chromatin remodeling. These are frequently all referred to as epigenetic mechanisms, though formally only mechanisms that are potentially heritable through cell divisions are considered epigenetic. Epigenetic changes in humans are typically not heritable through the germline, as epigenetic modifications are erased during gamete formation.
SLO 1: Understand the fundamentals of chromatin structure and remodeling.
Chromatin
Chromatin is a complex of DNA, proteins and RNA that allows chromosomes to be packaged and organized within a cell’s nucleus. Maintenance and remodeling of chromatin structure is essential for the proper regulation of gene expression, DNA replication, cell division and prevention of DNA damage. The fundamental unit of chromatin is the nucleosome, a structure comprised of 8 histone proteins around which DNA is wrapped.
Histones
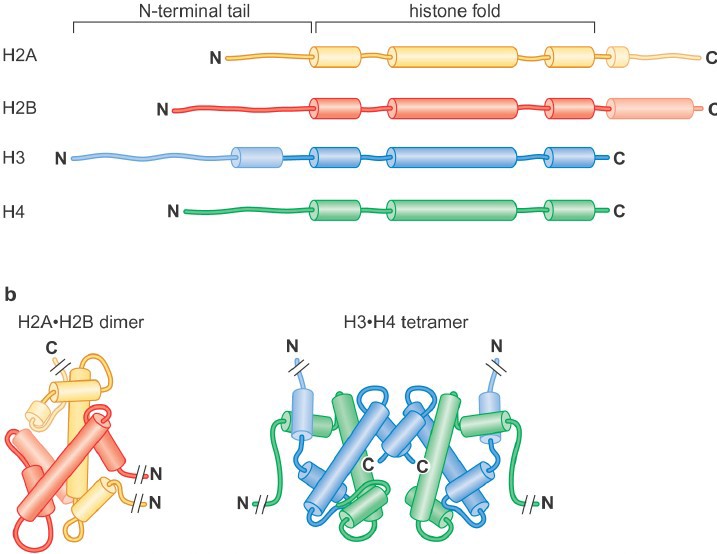
Histones are highly conserved, positively charged (lysine- and arginine-rich) proteins. All histones share a related histone fold domain comprised of 3 alpha helices, and the core histones also have additional C- and/or N- terminal extensions known as the histone tails.
There are four core his
tones: H2A, H2B, H3 and H4 (Figure 1). Two each of histones H3 and H4 form the H3-H4 tetramer, and H2A and H2B form dimers. One H3-H4 tetramer and two H2A-H2B dimers form the histone octamer (also called the “histone core particle.”.
The Nucleosome: the building block of chromatin
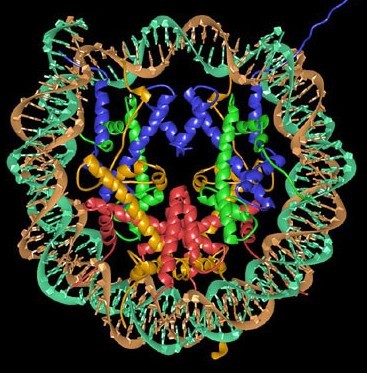
The nucleosome is formed when 146 bp of DNA wraps around the histone octamer (Figure 2), forming multiple ionic bonds between the DNA phosphate backbone and lysine and arginine residues in the histones. Linear DNA is wrapped around a succession of histone cores. DNA connecting adjacent nucleosomes is referred to as linker DNA. There is an average of ~50-60 bp of DNA in each linker region, though this can vary significantly at individual locations in the genome. Linker histones, such as Histone H1, interact with linker DNA to impact levels of chromatin folding.
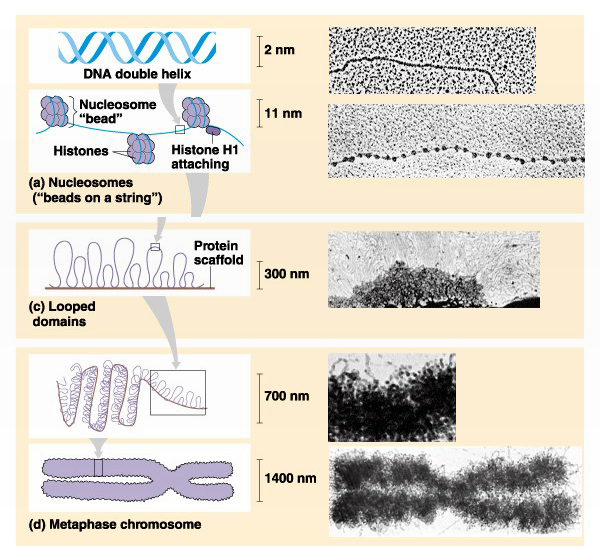
Chromatin folding
The nucleosome represents the first level of DNA packing, but in the nucleus DNA is condensed 10,000-50,000 fold over its fully extended length. An array of nucleosomes is also called a 10- nm fiber (its actual diameter is 11-nm). The 10-nm fiber is further folded via interactions with other proteins and association with an underlying proteinaceous scaffold that gives structure to both interphase chromatin (300nm) and the structure of metaphase chromosomes (700nm & 1400nm) (Figure 3).
ATP-dependent chromatin remodeling.
There is a large superfamily of ATP-dependent chromatin remodelers that use the energy of ATP hydrolysis to alter histone-DNA contacts, resulting in nucleosome sliding, histone or nucleosome
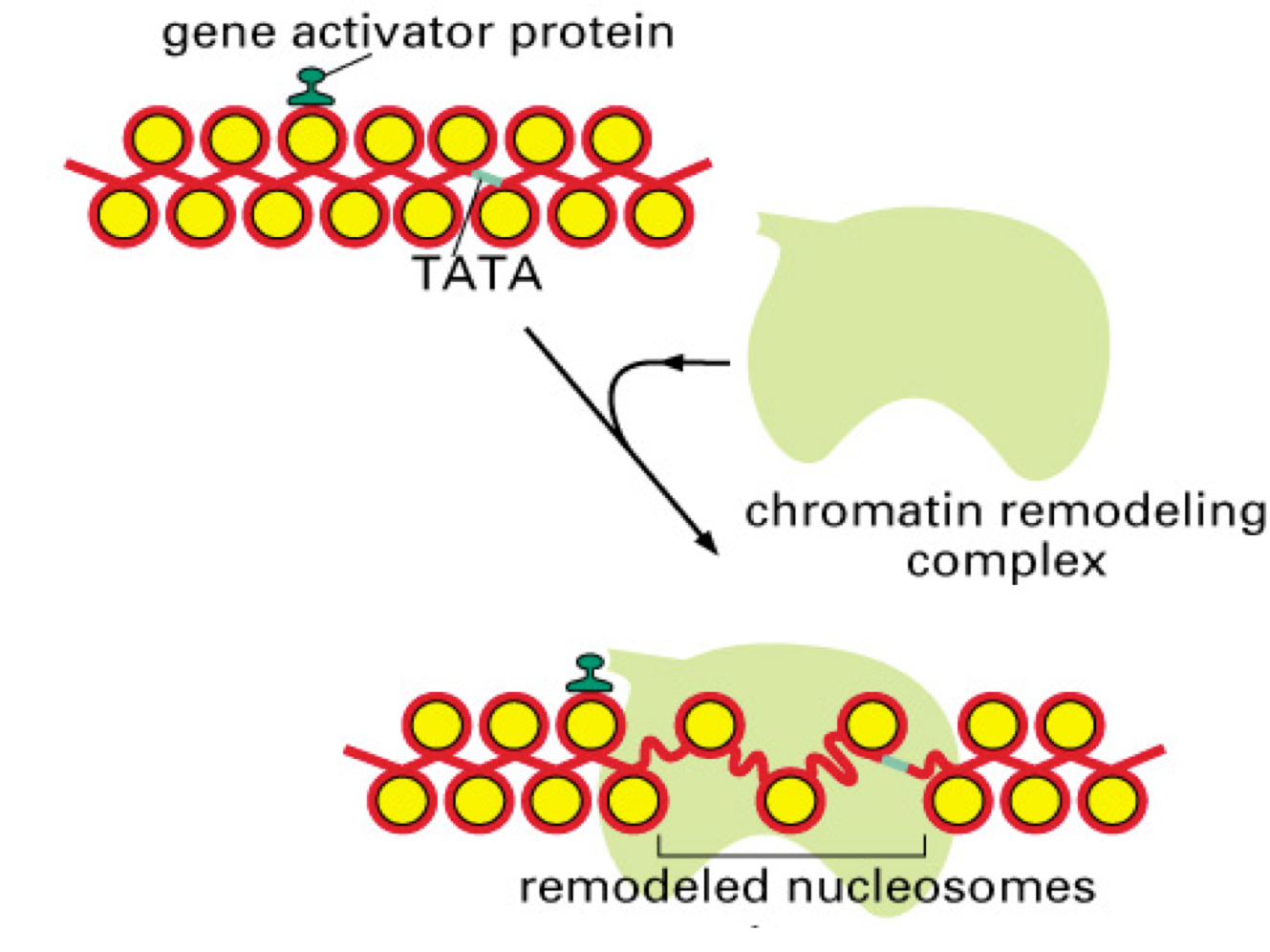
displacement from the DNA, and other alterations in nucleosome structure. The particular outcome depends on the remodeler involved and other protein factors acting at a particular site. Remodeling can lead to either accessible chromatin for gene activation (Figure 4), or can repress transcription (e.g. by moving nucleosomes to cover key promoter elements). There are numerous subfamilies within the superfamily of remodelers; the most important are the SWI/SNF, ISWI, CHD, and INO80 families. Most ATP-dependent remodelers exist in complexes of 2-20 subunits. Members of these complexes have been implicated in human diseases, including neurodevelopmental diseases and multiple cancers.
SLO 2: Describe the mechanisms by which covalent histone modifications and DNA methylation result in epigenetic regulation of gene expression.
Post-translational modifications of histones All histones have N-terminal tails, and some
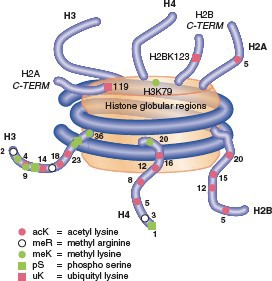
also have C- terminal tails. Tails are flexible extensions that
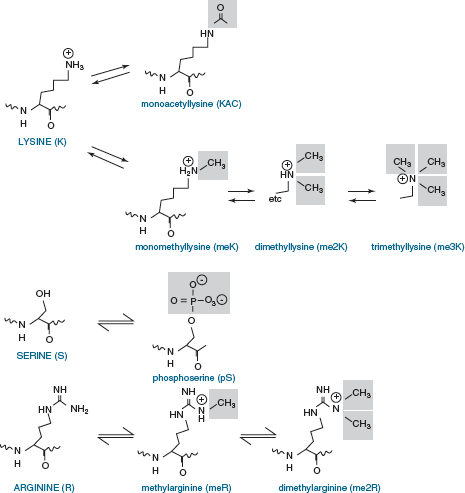
protrude fromthenucleosome.Multiplepost-translational modifications occur in the tails (Figure 5), though some also occur in the cores. The number of known covalent modifications is extensive and range from very large moieties such as ubiquitin or SUMO, to the small molecule modifications shown here. These include the best-characterized modifications, such as lysine acetylation, lysine and arginine methylation, and serine or threonine phosphorylation. Lysine can be mono-, di- or tri-methylated, and arginine can be mono- or di- methylated. Note that both phosphorylation and acetylation change the net charge, while methylation, even trimethylation, retains the positive charge on lysine and arginine (Figure 6).

Effects of histone modifications
Charge effects:
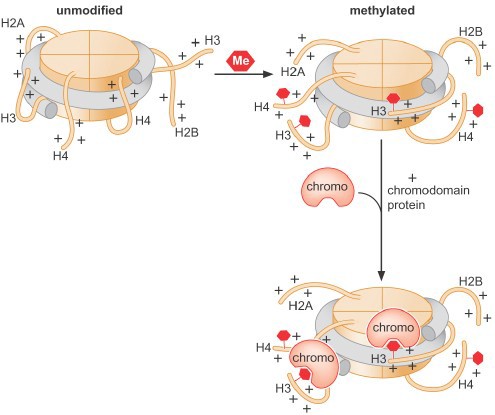
The positive charge on histone tails results in ionic interactions with the negative phosphate backbone of the DNA, both within a single nucleosome as well as between different nucleosomes in higher order structures. Acetylation of histone tails reduces these interactions and promotes unfolding of higher order structures and assists movement of the nucleosome along the DNA (Figure 7). Histone acetylation is associated with transcriptional activation.

Changestobindingsites:Post-translational modification of histone proteins can also produce novel binding sites for other proteins, which can result in a variety of functional outcomes. Enzymes that add epigenetic marks to chromatin are called writers. Proteins that recognize these marks are called readers. Enzymes that remove these marks are erasers. Note that these readers & writers are specialized: they recognize specific amino acid(s) within particular histone(s).
Histone methylation can be associated with either activation or repression, depending on the specific site of modification and thus the specific binding site generated (Figure 8). It is important to recognize that all histone modifications are reversible, but patterns of modificationmay be stably maintained through multiple cell divisions.
Some important classes of writers, readers and erasers:
Histone acetyltransferases (HATs) acetylate lysines (writer).
Histone methyltransferases (HMTs) methylate lysines or arginines (writer).
Bromodomains are common protein motifs that recognize acetylated lysines; they are commonly found in transcriptional activators, as well as in chromatin remodeling complexes (reader).
Chromodomains are one family of methyllysine binding domains, frequently found in repressive complexes (reader). Several other methyllysine and methylarginine binding motifs are known as well.
Histone deacetylases (HDACs) and histone demethylases remove acetyl and methyl marks, respectively (erasers). HDACs are generally found in repressor complexes. Histones can be extensively and simultaneously modified at numerous (different) sites within a single nucleosome. Modifications of the same residue are mutually exclusive (such as acetylation vs. methylation of the same lysine). The histone code hypothesis states that combinations of modifications define chromatin function and accessibility (Figure 9).
The “meanings” of many modifications have been defined, but the complexities of iinteractions of many modifications have yet to be clarified. Histone acetylation is
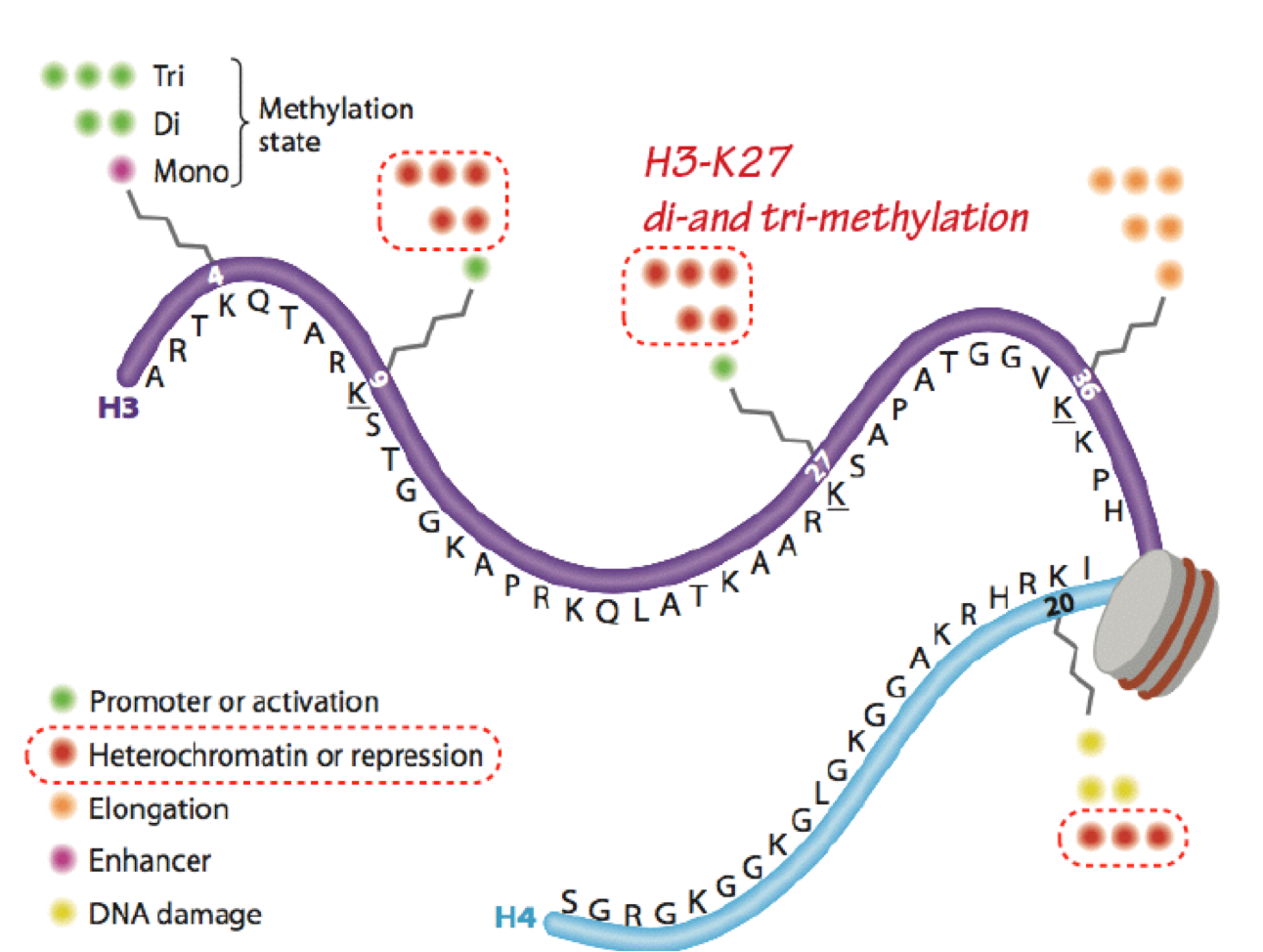
almost exclusively associated with transcriptional activation; while histone methylation is more frequently associated with transcriptional repression (eg H3- K27me2,3), some specific histone methylation sites are associated with activation (eg H3K4me2,3). Note that while the focus here is on transcription, histone modifications are also critically involved in all DNA transactions, including replication, recombination, and repair.
Post-translational modification of DNA
The most extensive (and best-studied) covalent modification of DNA is DNA methylation (Figure 10). It occurs on the 5C position of cytosine within CpG dinucleotides. (“CpG” is just a shorthand that specifies a 5’-CG-3’ sequence.) CpG dinucleotides are underrepresented (less frequent than expected by chance) in most of the genome, but enriched 100-fold in regions known as CpG islands.
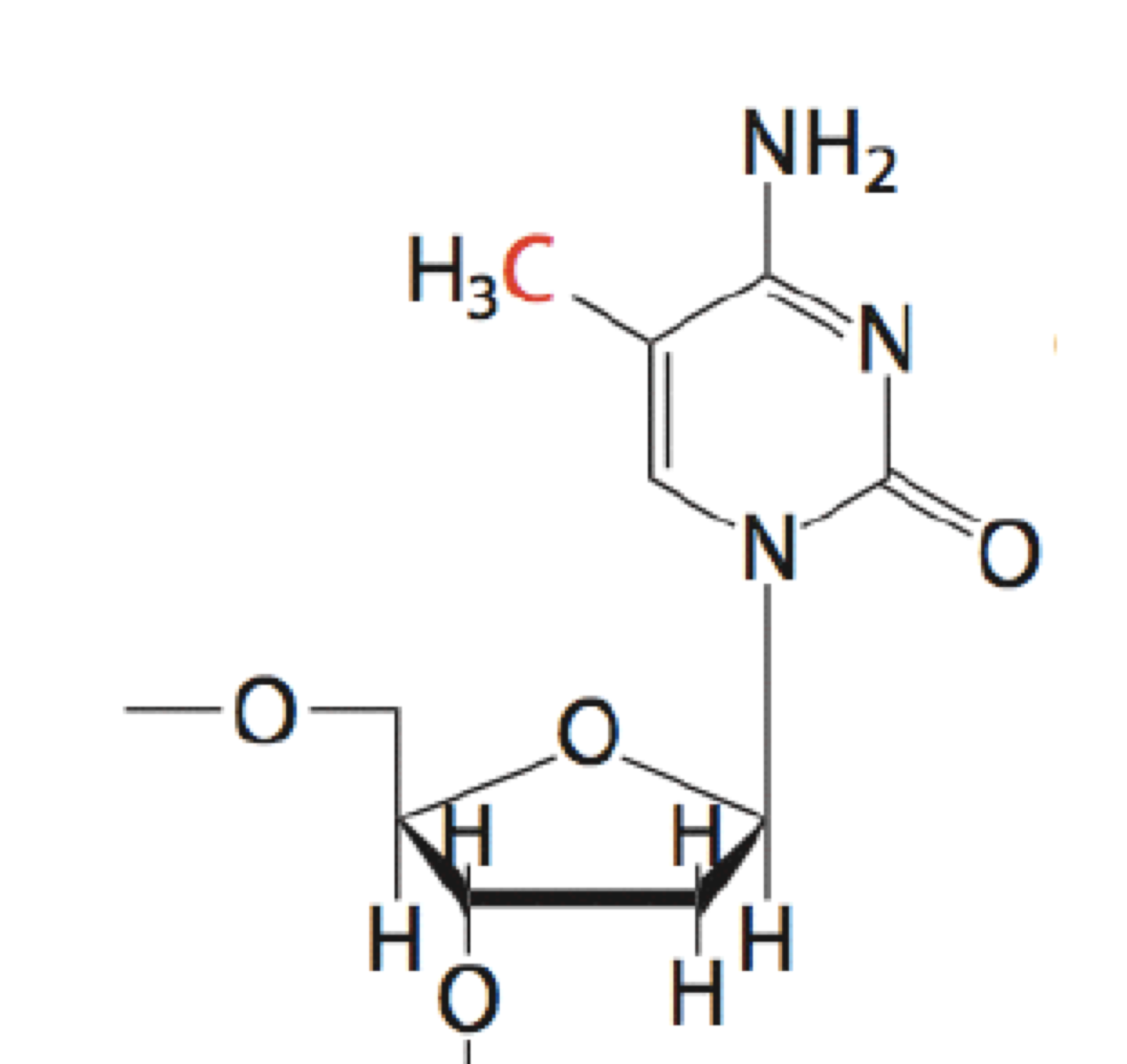
DNA methylation is typically a silencing epigeneticmark that is very stable through cell divisions. DNA methylation often occurs at CpG islands within gene promoter regions to silence gene expression. Commonly genes are progressively methylated during cell differentiation as genes whose expression is not needed for a particular cell type are silenced.
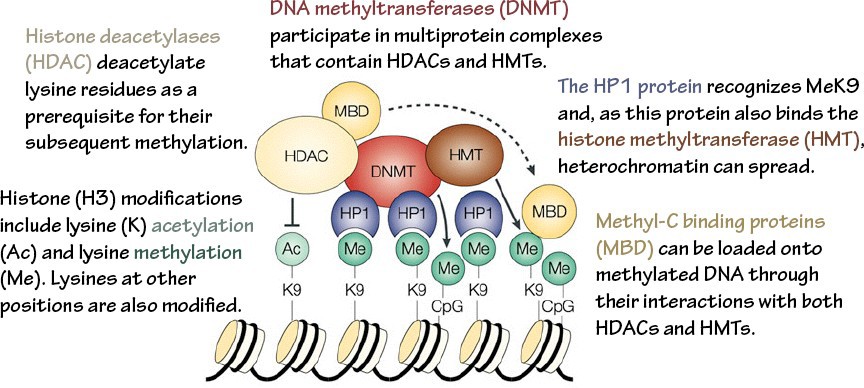
DNA methyltransferases (DNMTs) are the writers that perform this function. DNA demethylases are the erasers. Methyl-C binding domains (MBDs) are a common motif in readers of DNA methylation.
Gene silencing. Proteins that read and write repressive marks interact with each other, creating positive feedback loops that lead to sustained, self-reinforcing silencing (Figure 11). In addition, DNA methylation is maintained through replication with nearly 100% efficiency, ensuring methylation patterns are preserved through cell divisions. Thus, a fully silenced state is one of the most stable epigenetic states.
SLO 3: Demonstrate how epigenetic modifications result in imprinting and distinguish how imprinting errors lead to Prader-Willi or Angelman syndromes.
Imprinting: differential expression of paternal vs. maternal alleles.
- Imprinting is a specialized subtype of gene silencing. Patterns of allelic expression are predetermined based on the parent of origin of the allele. The allele from one parent is methylated and silenced (imprinted), while the allele from the other parent is active. Gender- specific patterns of imprinting occur during germ cell formation. First there is global demethylation followed by a sex-specific imprinting pattern that is laid down in primordial germ cells. These patterns are then maintained through fertilization and subsequent cell divisions (with the exception of
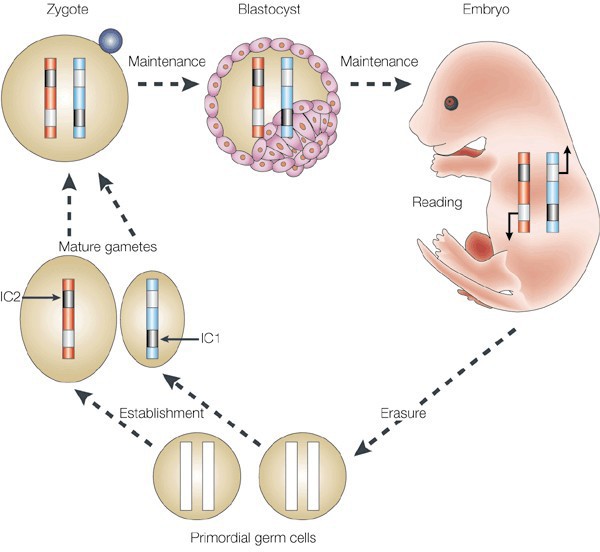
Figure 12. Schematic of parental imprinting. The epigenetic marks that drive parent-specific repression of certain genes (imprinting) are erased during the formation of primordial germ cells. Imprinting centers (ICs) then establish regions of repressed chromatin in the mature gametes that are unique to either the egg or sperm. These repressed regions are maintained in the fertilized zygote and throughout the lifetime of the resulting organism until those marks are once again erased in the new generation’s germline. some tissue-specific demethylation of certain genes) (Figure 12).
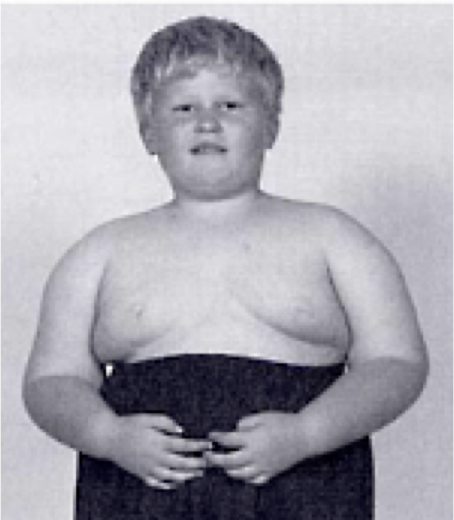
Diseases of imprinted loci. Several human syndromes result from mutations that impact imprinted regions. Two very different syndromes, Prader-Willi Syndrome (PWS) and Angelman Syndrome (AS), can actually be caused by the identicaldeletion in patients. PWS is characterized by chronic feelings of hunger and is the leading genetic cause of marked obesity; AS features severe motor and cognitive impairment. ~70% of patients have a 4-5 Mb deletion of a region known as the PWS-AS critical region (Figure 13). Which syndrome a patient exhibits is determined by which parent contributed the deletion. The typical deletion encompasses a large region with numerous imprinted genes, including two that are paternally imprinted (UBE3A and ATP10C) and multiple maternally imprinted genes.
 A
A
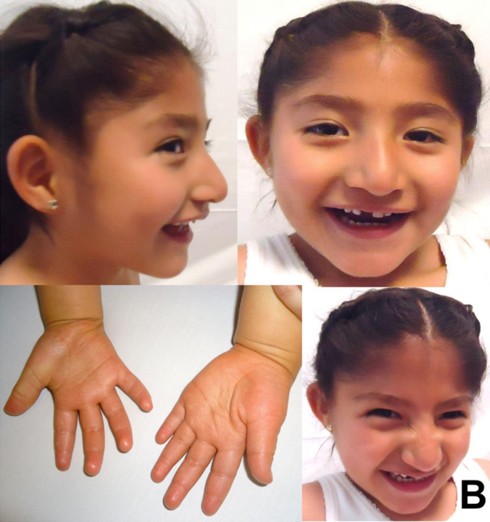
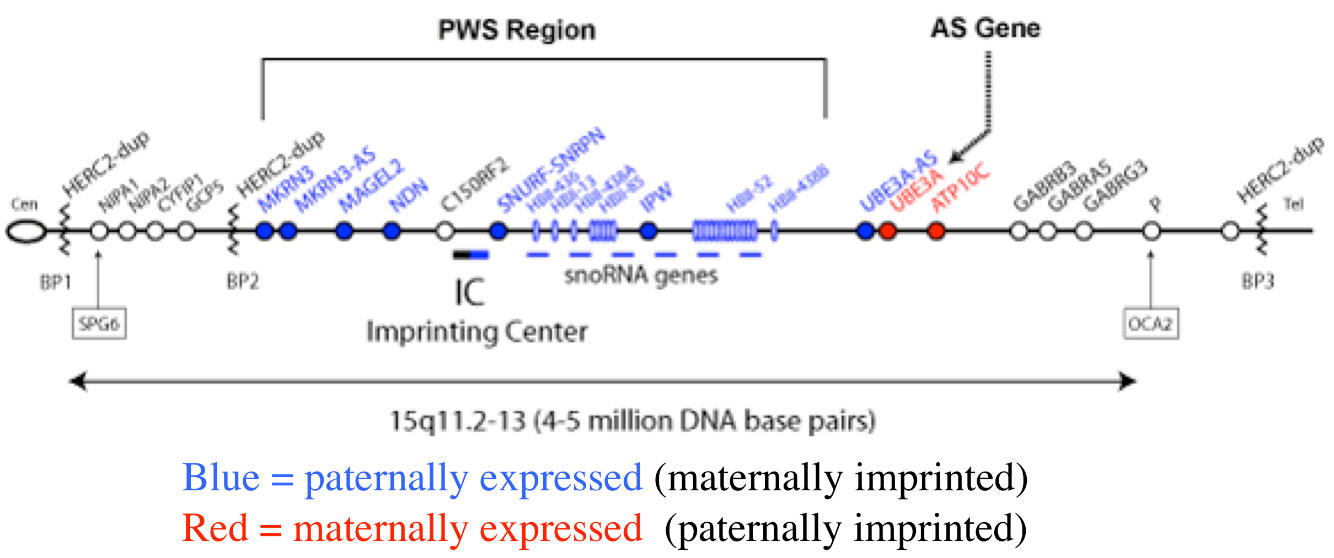
Prader-Willi syndrome results when the deletion comes from the father due to the loss of paternally expressed genes in the PWS critical region (i.e. none of the genes shown in blue in the figure are expressed, because they are imprinted in the intact maternal copy, and deleted in the paternal copy).
Angelman syndrome results when the deletion comes from the mother. In this case, it is the loss of the maternally expressed UBE3A gene (in the ubiquitin pathway) that results in the syndrome (mutations just in UBE3A alone also cause AS).
70% of the cases of both PWS and AS are due to deletion of the PWS-AS critical region. Both syndromes can also result from uniparental disomy, in which the individual inherited both copies of chromosome 15 from the mother (PWS, ~25% of cases) or the father (AS, ~7% of cases). In these cases, both chromosomes have the imprinting pattern of a single parent, and thus silence the same set of alleles on each copy. The remaining cases of PWS or AS result from mutations in genes that control imprinting, UBE3A mutations (AS), or other chromosomal rearrangements (Figure 14).
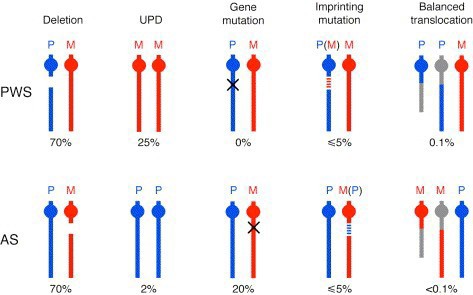
SLO 4: Illustrate how non-coding RNAs and covalent epigenetic modifications cooperatively regulate mammalian X inactivation.
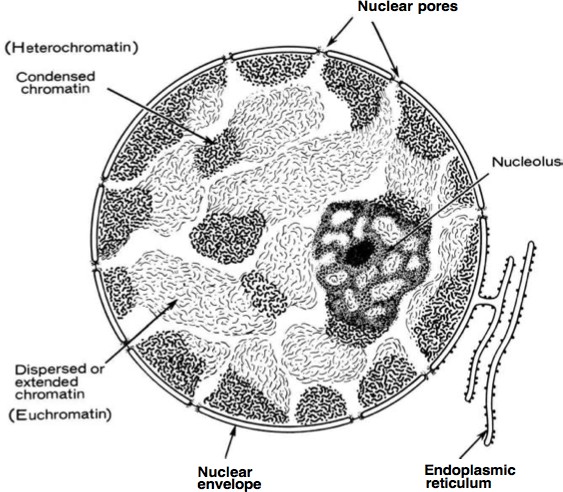
X-inactivation results in silencing of an entire chromosome. As described earlier, histone and DNA modifications can act in concert to establish stably silenced regions of the genome. Heterochromatin (Figure 15) is defined as highly compacted throughout the cell cycle, late replicating, and transcriptionally silent domains of chromatin. (All other regions are collectively known as euchromatin.) Some heterochromatic regions are always found as heterochromatin, such as telomeres and centromeres: these are constitutive heterochromatin. Other regions maybe euchromatic or heterochromatic depending on developmental stage, cell type, or other features. These are regions of facultative heterochromatin. The most dramatic example of facultative heterochromatin is the
silent X chromosome in female mammals.
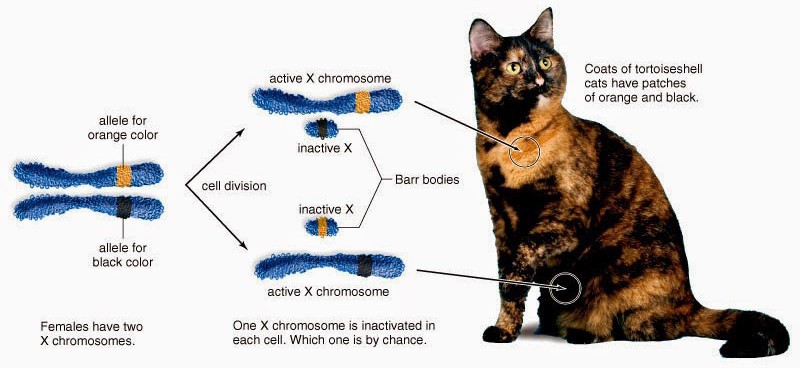
All female mammals are mosaic for expression of X chromosome genes, familiar to most in the coat color of calico cats (Figure 16). Because female mammals have two X chromosomes and males have one, the process of dosage compensation ensuresthat males and females receive the same levels of gene expression of X- linked genes. In mammals, dosage compensation is accomplished by [almost] entirely silencing one X chromosome in each cell in females. This silencing is random and occurs early in development; thus the adult is a patchwork of tissues in which cells were derived from an ancestor in which one of the two
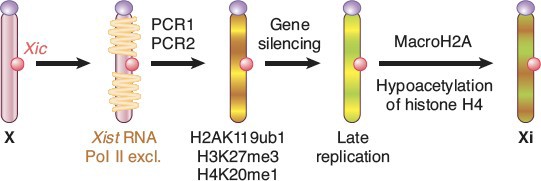
X chromosomes was silenced. The process was discovered by Mary Lyon and X chromosome inactivation is often referred to as “Lyonization.” The process of X-inactivation includes counting (all but one X will be silenced in individuals with >1 X), a choice where one X is designated to become the active X (Xa) and other(s) to become inactive (Xi), and finally the process of triggering extensive heterochromatin formation on Xi. Critical to this process are noncoding RNAs (ncRNAs) known as Xist and Tsix. Xist is a remarkable RNA that is transcribed stably from the Xic locus on the future Xi, and physically coats the chromosome to trigger the silencing process. Initially, both X chromosomes express Xist, which is destabilized by the presence of an antisense transcript from the same locus, Tsix. Selection of the future Xi involves inactivation of Tsix and stabilization of Xist. Xist accumulation on the Xi excludes RNA polymerase II, recruits silencing complexes, induces multiple histone modifications and inclusion of a variant of histone H2A found only on Xi, and finally DNA methylation of gene promoters on the Xi (Figure 17).
The role of X-inactivation in disease. Specific X-linked genes are mutated in many diseases such as Rett Syndrome, duchenne muscular dystrophy, color blindness and hemophila A. We typically think of X-linked conditions affecting males because they only have one X chromosome. Females, however, may be heterozygous for the mutation. In other words they may have one mutated and one normal allele, although any given cell expresses only one allele because of X-inactivation.
Females can be affected by X-linked mutations as a result of skewed Lyonization, but may have distinct clinical phenotypes because some of their cells are normal, while other cells express the mutated gene on the X chromosome. For example, young girls with pathological mutations in one copy of the X-linked Methyl CpG-binding Protein gene (MECP2) present with Rett Syndrome, an autism-like neurodevelopmental disorder with an onset between 6-18 months. In contrast, boys with MECP2 mutations, who survive to term, present with different phenotypes including generalized mental retardation. Aside from being located on the X- chromosome, the MECP2 gene produces a protein product that binds to methylated DNA throughout the genome to regulate transcription. Rett Syndrome, therefore, is not only influenced by the epigenetics of X-inactivation, but also affects the epigenetic state of the cell through the misregulation of expression of methylated DNA. The severity of an X-linked condition will depend on the pattern of X- inactivation. Just as calico cats can have coat color spots of variable size and coverage, X- inactivation in humans can be skewed for a given body region or organ system.
Feedback: