
10 DNA Replication and Repair
Session Learning Objectives:
- Illustrate DNA replication and identify proteins that are targets for inhibiting DNA replication.
- Explain why telomere replication presents special problems and the disorders that could develop if defective, such as dyskeratosis congenita.
- Describe the major sources of DNA damage and errors and the pathways used to recognize and correct these errors.
- Analyze how defects in different DNA repair pathways lead to specific syndromes, including cancer-predisposition syndromes: Li-Fraumeni syndrome, Lynch syndrome, Xeroderma pigmentosum, Ataxia telangiectasia and hereditary breast and ovarian cancer (HBOC) syndromes.
- Describe how DNA repeat expansion relates to the presence and severity of specific disorders: Fragile X syndrome/Fragile X-associated tremor/ataxia syndrome (FXTAS), Huntington disorder, myotonic dystrophy.
- Describe how repeated DNA sequences and homologous recombination contribute to the appearance of interstitial deletion syndromes.
SLO 1. Illustrate DNA replication and identify proteins that are targets for inhibiting DNA replication.
DNA replication
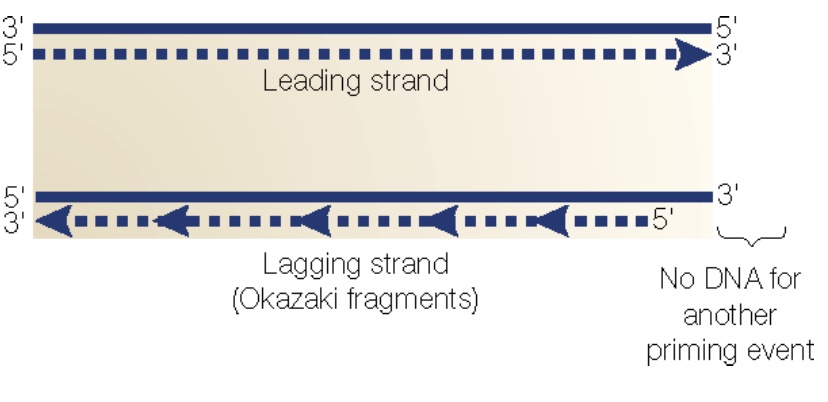
DNA replication is semiconservative: Each daughter strand contains one of the original parent strands and one new strand. The template strands are antiparallel, and polymerase can only synthesize DNA in one direction (5´→ 3´). The fork moves in one direction (i.e. direction of DNA unwinding; green arrow), while replication of each strand is in the opposite direction—yet still simultaneous. The strand that is being synthesized in the same direction as fork movement is the leading strand, and it is synthesized continuously as the fork unwinds. The lagging strand is synthesized as a series of short Okazaki fragments. Each Okazaki fragment requires a new primer. Primase is the complex of enzymes that makes these primers.
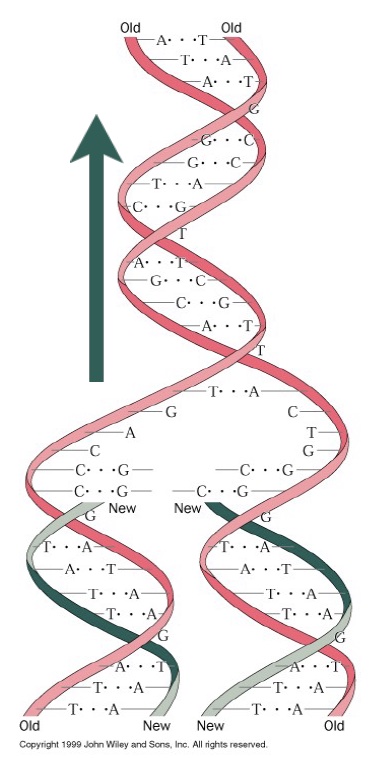

DNA Polymerases require an initiating primer made from either DNA or RNA. RNA polymerases do not. Primase is a special DNA-dependent RNA polymerase that synthesizes primers for the leading strand. A DNA polymerase extends these primers, then the main replicative DNA polymerases synthesize the leading and lagging strands. During ongoing lagging strand synthesis, the primers are removed, any gaps between the Okazaki fragments are filled in by DNA polymerase, and finally nicks are sealed by DNA ligase.
Other essential components of replication:
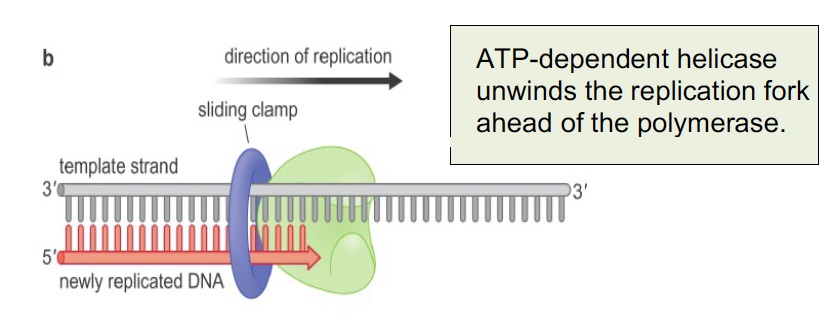
The sliding clamp is a processivity factor (keeps polymerase complex from falling off the DNA). The human sliding clamp is PCNA (Proliferating Cell Nuclear Antigen), which is often used in histology as a marker of DNA synthesis and cell proliferation. It is a trimeric complex assembled around the DNA by an ATP-dependent clamp loader on the leading strand and at each new Okazaki fragment.
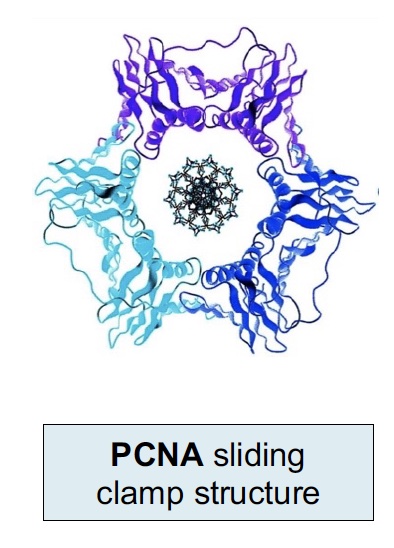
Another marker for cell proliferation is Ki-67 (MKI67) (not shown here and not to be confused with PCNA). Ki-67 is associated with the transcription of ribosomal RNA in the nucleus and is present throughout an active cell cycle. Ki-67 is increased during the S phase where DNA is being synthesized.
All of these key activities are shown in the figure below showing proteins at the replication fork; the inset shows how the leading and lagging strands are coordinated. As each Okazaki fragment is synthesized, the loop grows until the polymerase encounters the 5’ end of the prior Okazaki fragment. Then the loop is released, a new clamp is loaded, and the next fragment begins.

SLO 2. Explain why telomere replication presents special problems and the disorders that could develop if defective, such as dyskeratosis congenita.
Topoisomerases relieve the superhelical tension (positive supercoiling) that occurs ahead of a moving replication fork, by breaking the DNA to relieve the torsion, then resealing the break.
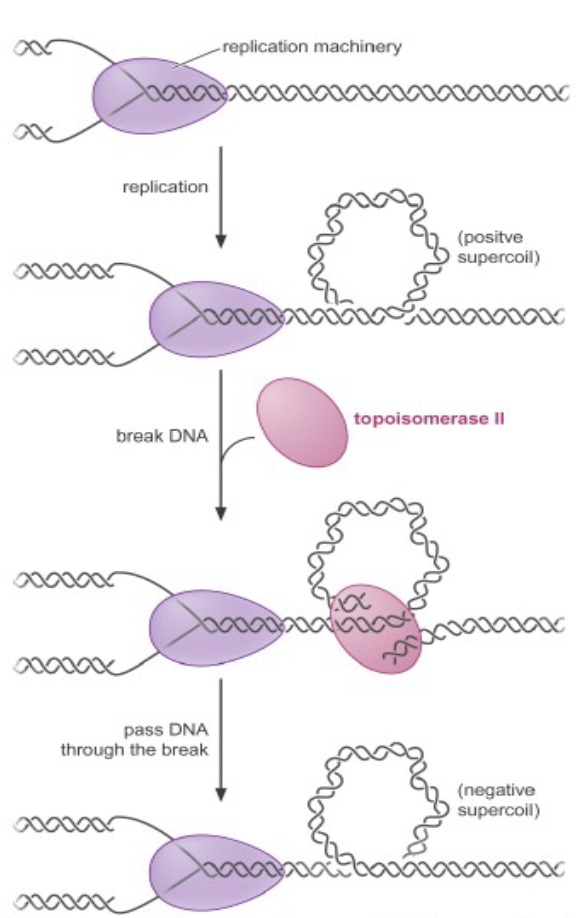
Topoisomerase inhibitors are powerful anticancer drugs and antibiotics. For example, camptothecin-derived compounds (like Topotecan, used for ovarian & lung cancer) block replication by converting the topoisomerase reaction into a dead-end reaction with the topoisomerase covalently linked to broken DNA. This results in genome fragmentation and cell death.
Telomerase dysfunction results in premature death of tissues that rely heavily on stem cell divisions. Mutations in TERT, TERC and several other genes cause the genetic disorder Dyskeratosis congenita (DC), which results in irreversible degeneration of skin tissue, early hair graying and loss and bone marrow failure, among other clinical presentations.
Chromosomes are replicated by many polymerase complexes, operating in parallel. Each chromosome has many replication origins. At each origin a replication bubble opens by melting the DNA duplex. Within each bubble, two replication forks move away from the origin, each with a leading and lagging strand. Replication terminates when forks collide or reach the end of the chromosome. This creates two problems.

Problem one: How, with so many origins (that fire at different times during S phase), can you ensure any given origin initiates once and only once per cell division cycle?
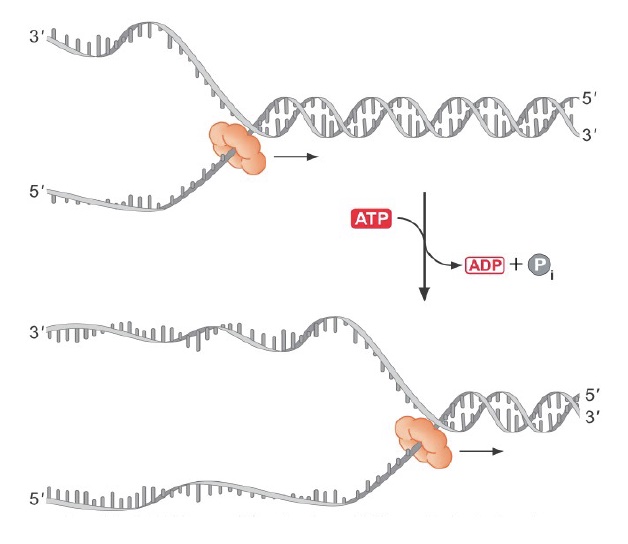
ORC (Origin Recognition Complex) binds to origins throughout the cell cycle, marking each initiation site. ORC then brings in additional proteins that load the MCM complex (Panel A and B in figure). MCM is the replication helicase, and similar to the sliding clamp, encircles DNA and can’t fall off.
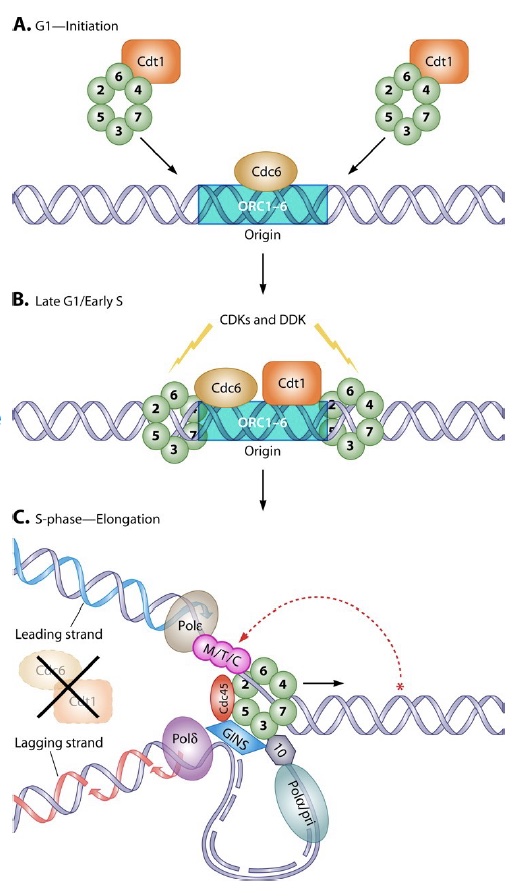
When the signal arrives to initiate replication (a cascade of phosphorylation events), the MCM helicase is activated to fire the origin, and MCM loading proteins (Cdc6/Cdt1) are destroyed to prevent re-licensing! (Panel C in figure). They will not return until the G1 phase of the cell cycle.
Problem 2: The “end replication problem.” At each end of the chromosome, replication results in a 3’ overhang because after removal of the endmost RNA primer on the lagging strand, there is no upstream sequence to prime the DNA polymerase filling in. Thus, chromosomes shorten with every replication cycle!
The solution: Telomeres and telomerase.
Telomerase is made of two components:
TERT, an RNA-dependent DNA polymerase (aka, reverse transcriptase)
TERC (Telomerase RNA component or hTR), an RNA molecule that provides the template for the telomeric repeat sequence.
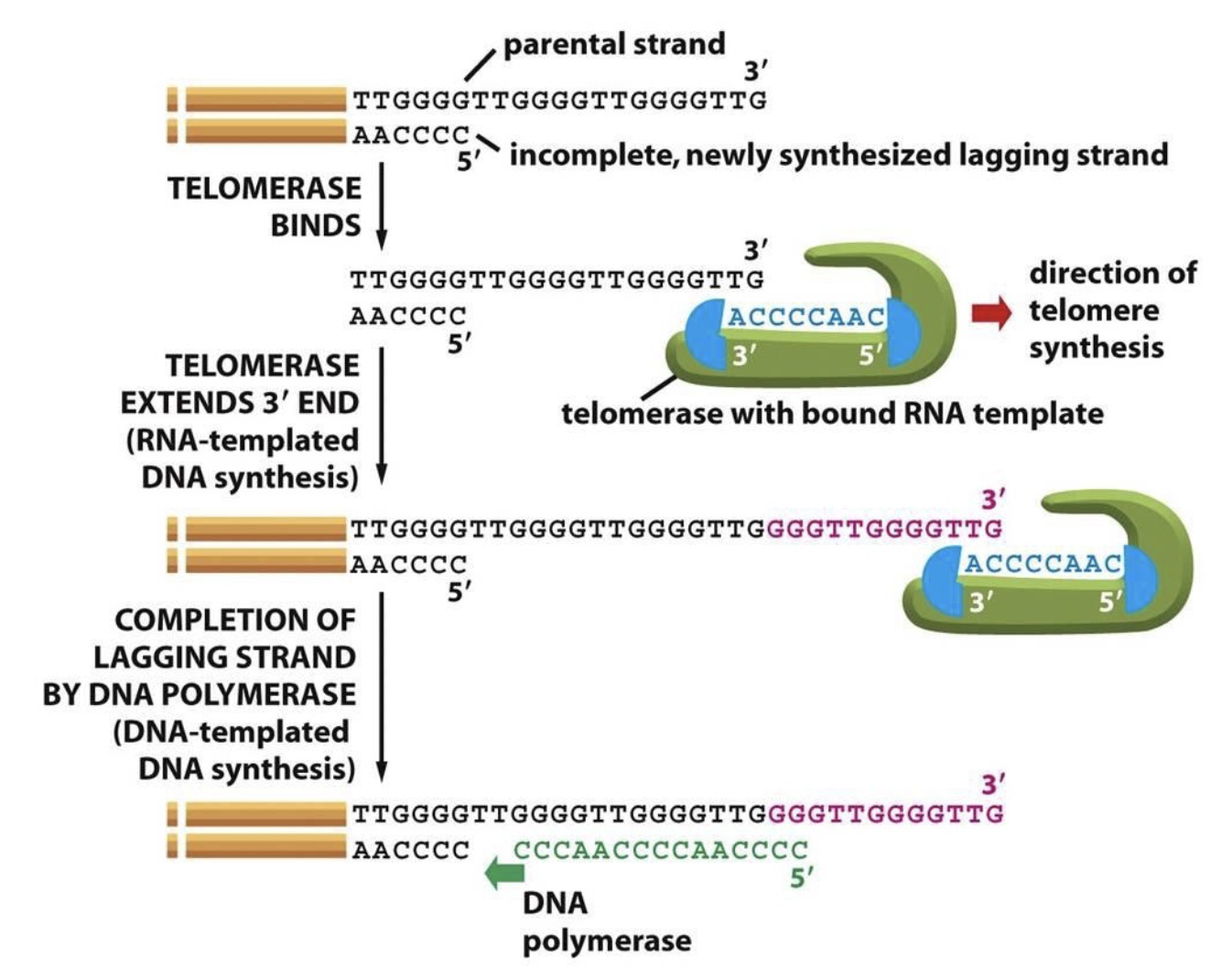 Telomerase is active during embryogenesis and in stem cells, but not in more differentiated cells. Thus, telomeres shorten with each cell division in these cells. (Cancer cells frequently reactivate telomerase to achieve replicative immortality.) Telomerase synthesis involves binding to the 3’ end of the G-rich telomeric parental strand and aligning with the complementary TERC RNA template. TERT then adds 6 deoxynucleotides using the RNA template and translocates to the new 3’ end of the DNA to repeat the process.
Telomerase is active during embryogenesis and in stem cells, but not in more differentiated cells. Thus, telomeres shorten with each cell division in these cells. (Cancer cells frequently reactivate telomerase to achieve replicative immortality.) Telomerase synthesis involves binding to the 3’ end of the G-rich telomeric parental strand and aligning with the complementary TERC RNA template. TERT then adds 6 deoxynucleotides using the RNA template and translocates to the new 3’ end of the DNA to repeat the process.
SLO 3. Describe the major sources of DNA damage and errors and the pathways used to recognize and correct these errors.
SLO4. Analyze how defects in different DNA repair pathways lead to specific syndromes, including cancer-predisposition syndromes: Li-Fraumeni syndrome, Lynch syndrome, Xeroderma pigmentosum, Ataxia telangiectasia and hereditary breast and ovarian cancer (HBOC) syndromes.
DNA repair
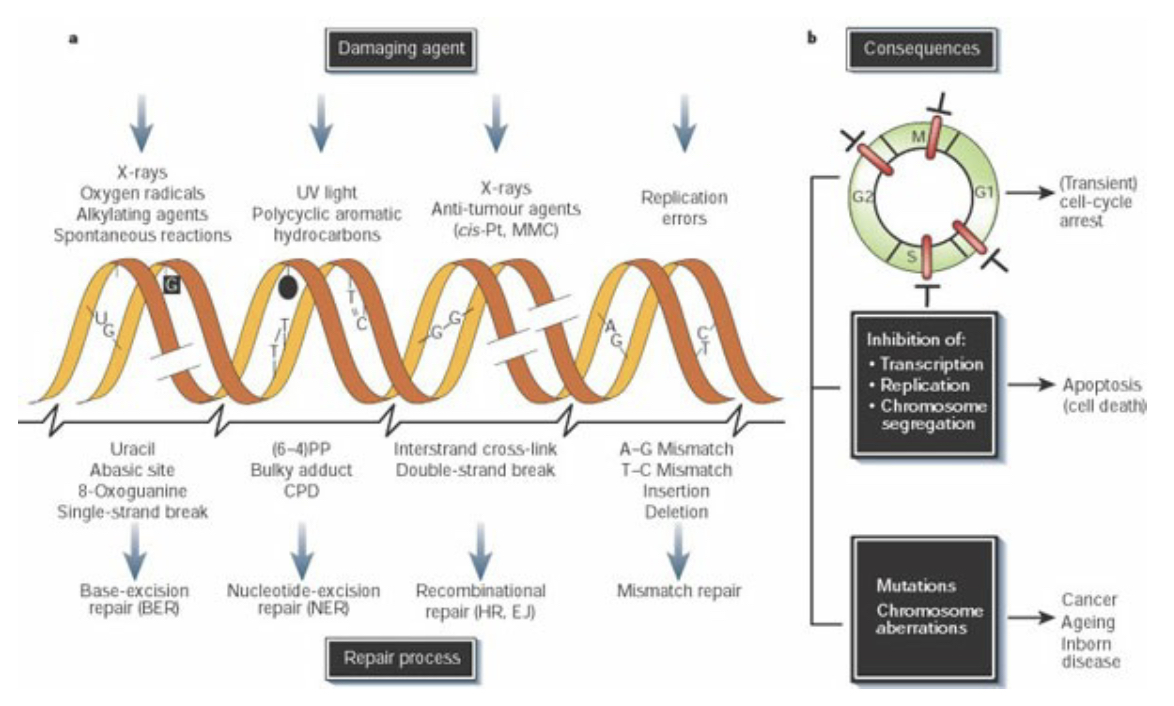
Cells are subjected to many sources of DNA damage, both extrinsic (such as UV or ionizing radiation) and intrinsic (such as errors of replication, endogenous reactive oxygen species, and spontaneous lesions). Different sources of damage create different types of DNA lesions, which in turn are recognized and repaired by specialized repair pathways. DNA damage triggers cell-cycle arrest (checkpoint activation), which will be maintained until either the damage is repaired or the persistence of damage triggers apoptosis. Unrepaired damage can result in fixation of a mutation, from single base pair changes to major chromosomal rearrangements.
DNA Repair pathways
DNA repair pathways fall into several broad categories: direct reversal, in which damage is directly reversed in situ with no cleavage of the DNA backbone; excision repair pathways (BER, NER, MMR), in which a single strand of DNA containing damage is excised (cleaved 5’ and 3’ of the damage and removed) and replaced by DNA polymerases using the undamaged complement as a template; and double strand break repair (DSBR) pathways that contend with damage resulting in breakage of both strands of DNA.
Only one type of direct reversal exists in human cells: direct removal of alkylation damage. Base alkylation, most commonly methylation or ethylation, usually occurs due to exposure to alkylating drugs such as those used in chemotherapy.
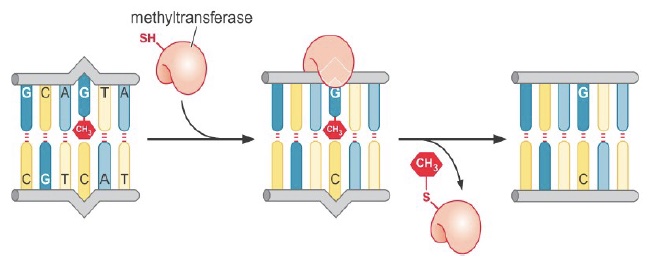
Repair methyltransferases transfer the alkyl group directly to a cysteine residue in the repair protein. This is a suicide repair mechanism, as the alkyl group is then permanently covalently attached to the protein.
Base excision repair (BER)
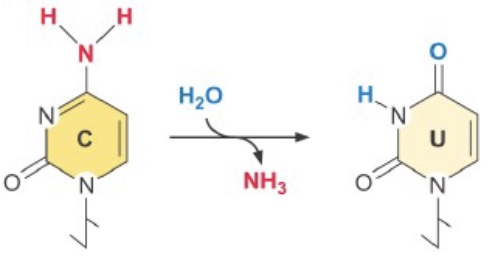
One common source of DNA damage in the normal cellular environment is spontaneous deamination of cytosine—it occurs at ~100 C’s/cell/day! Cytosine deamination creates uracil, easily recognized as an error. However, deamination of 5-methyl cytosine yields thymine and causes a T-G mispairing. [This may be part of the reason CpGs are relatively rare outside of CpG islands—it is harder to repair accurately. To compensate, repair of T-G mispairs outside of replication are biased toward replacing the T.]
Spontaneous depurination (loss of the adenine or guanine base from the deoxyribose sugar) is even more frequent, at ~5000 events/cell/day.
Both of these types of damage, as well as some other small base lesions, are repaired via base excision repair (BER). First, a damaged base is released from the deoxyribose by the action of a glycosylase enzyme, creating an abasic site (not necessary for spontaneous depurination). Then, an AP (apurinic/apyrimidinic) endo/exonuclease cleaves the phosphate backbone on either side of the abasic site. A DNA polymerase inserts the correct base(s) and ligase seals the final nick.
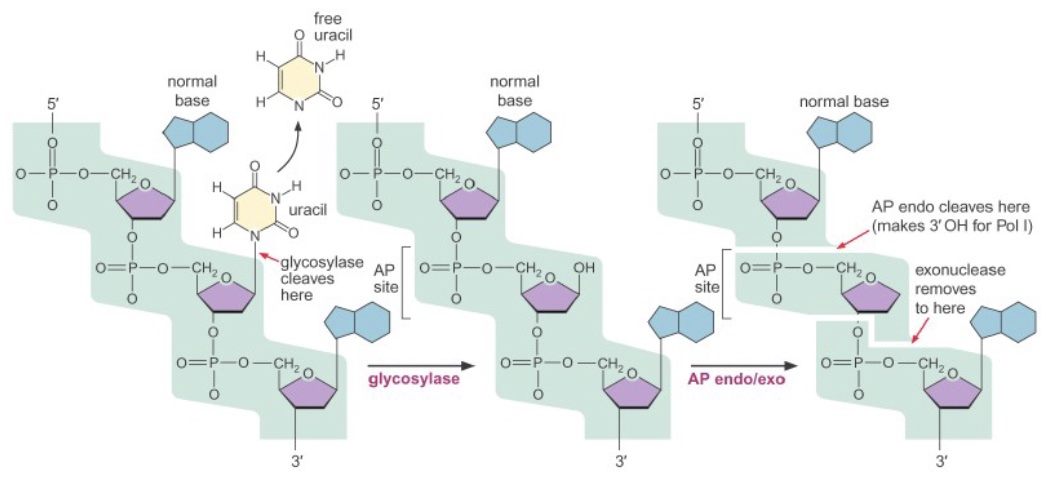
Nucleotide excision repair (NER)
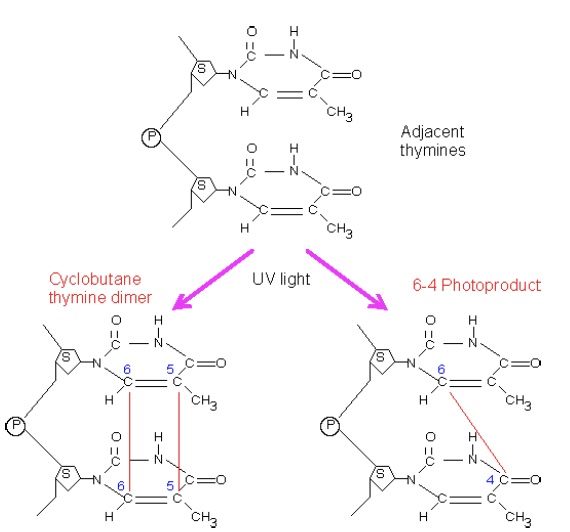
The NER pathway primarily recognizes so-called “bulky lesions,” lesions that create significant distortions in the structure of the DNA double helix. One of the most important of these is damage resulting from UV light exposure, which causes dimerization of adjacent pyrimidines in one strand. This dimerization can involve one bond (forming a 6- 4 photoproduct, 6,4-PP) or two bonds to create a cyclobutane pyrimidine dimer, CPD.
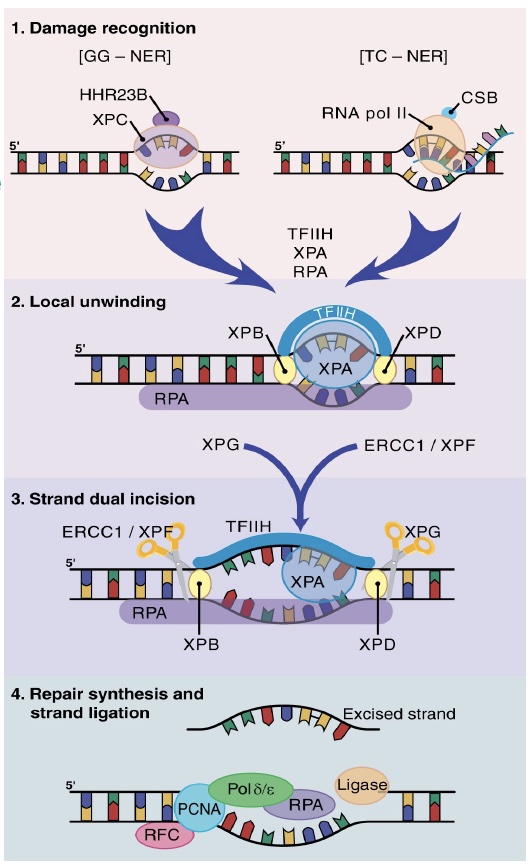
NER is subdivided into two pathways based on the initial recognition of damage. In global genome (GG)-NER, a helix-distorting lesion is detected anywhere in the genome by damage surveillance proteins such as XPC. In transcription-coupled (TC)-NER, damage is recognized when it causes an elongating RNA polymerase II to stall.

Mismatch repair (MMR)
Replication errors occur at ~1 base change/cell division missed by DNA polymerase’s proofreading capacity (without proofreading the error rate would be more like 100 bases/cell division). The mismatch repair system operates in conjunction with replication to repair remaining errors. The human MutS (hMutS) complexes recognize the mismatches. hMutS-alpha (a dimer of MSH2 and MSH6) recognizes single mismatches, while human hMutS-beta (MSH2-MSH3) recognizes insertion/deletion loops resulting from replication slippage. The hMutL-alpha complex (MLH1 and PMS2) links hMutS to the replication machinery and can nick the DNA, although usually a preexisting nick between Okazaki fragments is used as an excision point. Replication machinery fills in the remaining gap after excision.

Inherited mutations in MMR machinery (most often in MSH2 and MLH1) result in Lynch syndrome (previously known as hereditary nonpolyposis colorectal cancer, HNPCC), a genetic syndrome with a dominant pattern of inheritance that results in a high risk of colon cancer, endometrial cancer, and several others. A hallmark of Lynch syndrome is microsatellite instability (MSI), caused by the failure to correct replication slippage at repeated sequences, resulting in changes in repeat numbers (increases or decreases) in tumor cells. Clinical testing of tumor tissues may include immunohistochemistry and PCR-based microsatellite instability analysis. However, a definitive diagnosis of Lynch syndrome requires identification of a mutation in one of the four human mismatch repair genes, MLH1, PMS2, MSH2 and MSH6, or potentially an inactivating methylation of one of these genes.
A newer use of identifying defects in MMR genes is in evaluating tumor mutational burden (TMB) (number of somatic mutations per megabase.) The efficacy of treatment with immune checkpoint inhibitors for multiple types of cancers may be estimated by evaluating the TMB, which will be elevated in tumors harboring defects in mismatch repair leading to microsatellite instability or defects in other DNA repair genes. Current research is directed at evaluating the predictive value of high TMB on increased long-term survival of patients with different tumor types.

Double strand break repair (DSBR)
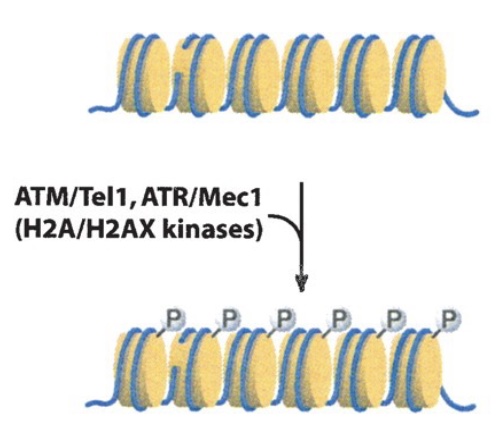
One of the most serious categories of DNA damage is DNA double-strand breaks (DSBR). A single break will arrest the cell cycle until repair is completed or the cell undergoes apoptosis. DSBR is divided into two subcategories: non-homologous end joining (NHEJ) and homologous recombination (HR). Both pathways involve immediate and extensive phosphorylation of the histone variant H2AX at the site of damage (and over megabases surrounding the damage site) by the checkpoint kinases ATM/ATR. This phosphorylation is involved in subsequent interactions with repair factors.
In contrast, HR (which has many subtypes) is a highly accurate repair system that uses homologous sequence (usually a sister chromatid) as a template for accurate repair of a damage site. Repair by HR counterintuitively begins with 5’ resection of the broken ends to generate long 3’ overhangs. These overhangs are then used to invade the homologous donor sequence, and DNA polymerase then extends from the 3’ end using the homologous donor as a template.
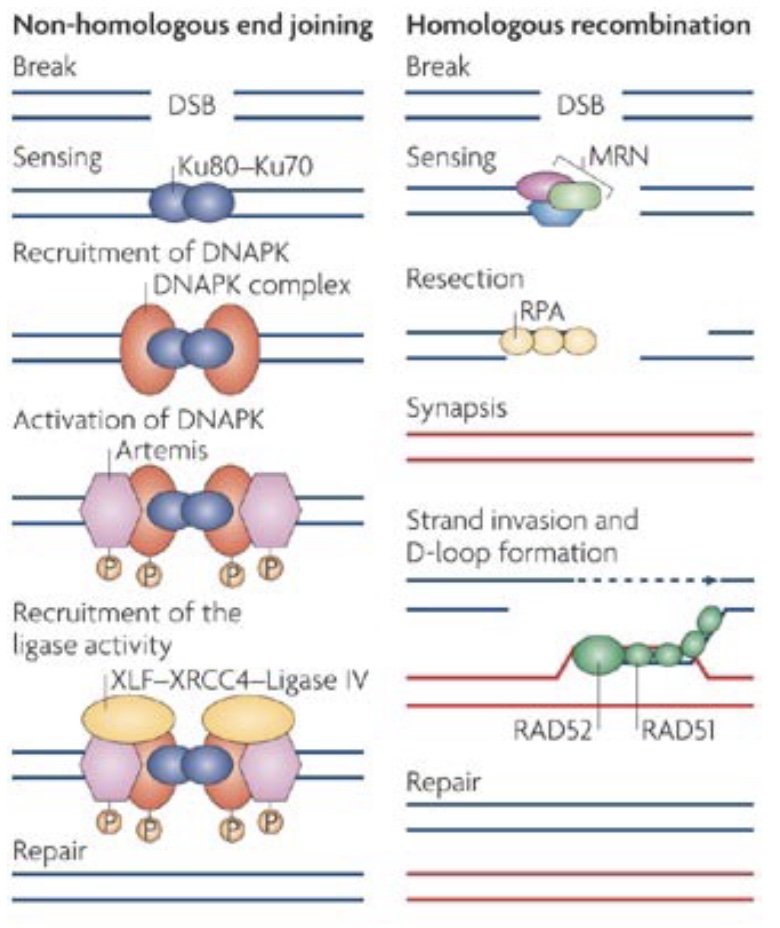
Like NHEJ, HR is a repair system that is also used for another cellular process: meiotic recombination during gametogenesis. During meiosis, DSBs are deliberately induced, and then HR machinery produces meiotic recombinants. Errors can occur during this normal meiotic recombination process as well, which can lead to significant chromosomal aberrations, such as interstitial deletions causing microdeletion syndromes, discussed further later in this chapter.
SLO 5. Describe how DNA repeat expansion relates to the presence and severity of specific disorders: Fragile X syndrome/Fragile X-associated tremor/ataxia syndrome (FXTAS), Huntington disorder, myotonic dystrophy.
Trinucleotide Repeat Disorders
Some genetic disorders arise from a progressive expansion of a region of DNA containing multiple 3-nucleotide repeats (triplet repeats). The phenomenon of potential trinucleotide repeat expansion, from one generation to the next, is refered to as “genetic anticipation.” In these disorders, healthy individuals have a variable number of repeats below a particular threshold; repeats beyond that threshold number present with a disorder, with the severity of the disorder typically correlated with the number of repeats (but there are exceptions). Repeat expansions are caused by strand slippage as discussed above in the context of mismatch repair. Individuals with repeats in the normal range are not at any increased risk of their offspring developing disease, but individuals in the premutation range have repeats approaching the threshold which are unstable and more likely to expand (or rarely, contract) in the next generation. Remarkably, the sex of the parent can affect the risk of repeat expansion.
Fragile X syndrome (FXS) is the second most common inherited form of intellectual and developmental disability (after Down syndrome). Fragile X syndrome is visible cytologically in affected patients as a constricted site in the X chromosome that appears “fragile” but is not (located at Xq27.3). The repeated sequence is a CGG triplet in a non-coding 5’ untranslated (5’UTR) region of the FMR1 (Fragile X Messenger Ribonucleoprotein 1) gene. The repeats may interfere with gene function by providing a substrate for CpG methylation and silencing of FMR1 gene transcription or may involve other mechanisms such as hybridization of the CGG region in the mRNA to the complementary DNA to form an RNA-DNA hybrid mediating epigenetic gene silencing. The FMRP protein encoded by this gene is normally involved in translation and trafficking of mRNAs in neurons and plays a role in learning and memory.
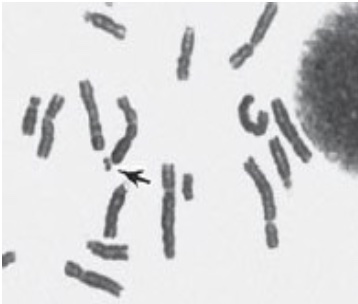

Disease severity generally correlates with CGG trinucleotide repeat length and ranges from 55-200 repeats for the premutation alleles and >200 to several hundred to several thousand repeats for the full mutation, as compared with 5-44 CGG repeats in a normal allele. Because the disorder is X-linked, more males are affected than females, however females heterozygous for a full mutation are also at risk for intellectual disability. X-inactivation in the female will play a role in presentation of the disorder, depending on which X is inactivated in which tissues. In addition to mild to moderate intellectual disability, males typically present with characteristic facies of a long narrow face, large ears and macroorchidism (large testes). Males with premutations may develop an adult-onset related neurodegenerative disorder called Fragile X tremor-ataxia syndrome (FXTAS).
An allele in the premutation range may be stable for generations or increase in size when inherited from the mother (or rarely regress). However, sperm carry only premutation alleles, even if a male has a full mutation. Consequently, males with a premutation or full mutation pass the premutation to all of their female children (and of course not to their male children, who do not inherit the father’s X chromosome.) Furthermore, in father-to-daughter transmission, the size of a premutation contracts in about one third of female offspring. The mechanism underlying these observations has not been established.
Myotonic dystrophy (aka dystrophia myotonica, DM) is one of the most common inherited forms of progressive muscle disease. Age of onset decreases and severity increases with larger repeat sizes. The image to the right shows three generations of individuals with DM: the infant has >1000 repeats, and the mother and grandmother have ~100.

There are two types of autosomal dominant DM (1 and 2), both caused by repeat expansions in the transcribed portion of the genes. DM Type 1 is caused by the expansion of the CTG triplet repeat in the 3’-UTR of the DMPK gene, encoding a protein kinase that is important in muscle, heart and brain, though its precise role is not clear. Type 2 is milder and is caused by a tetranucleotide repeat expansion in the first intron of the CNBP gene. In both types of DM, the pathology appears to be primarily due to the excessively long mRNAs produced, which form inclusions that interfere with cellular translational machinery and/or other functions.
Huntington Disorder (Huntington Disease) (HD) is an autosomal dominant progressive disorder causing cognitive, motor, and behavioral changes. The mean age of onset is 35-44 years (with earlier onsets with increased repeat lengths) with a survival time of 15-18 years from onset. The earliest manifestations are usually subtle motor defects including involuntary movements (chorea) and irritability. Cognitive decline ultimately leads to dementia.

The repeated sequence is a CAG codon for glutamine in the first exon coding region of the huntingtin (HTT) gene. The resulting Huntingtin protein contains a polyglutamine (polyQ) tract. Huntingtin is a poorly understood, ubiquitously expressed protein (highest expression in brain and testes) with multiple cellular functions. It is not clear which loss of function might contribute to HD. However, one key feature of the polyQ tracts is that turnover of mutant huntingtin (mHTT) leads to accumulation of undegraded polyQ fragments, which, because of their polar nature, tend to form aggregates (with mHTT and other proteins), leading to large inclusions in neurons. One of the proteins that becomes trapped in these inclusions is histone acetyltransferase CBP (which itself has an 18-Q tract), which is a coactivator for many genes. This trapping of CBP has been implicated in the neural pathology of HD, however there are other poly Q proteins, such as transcription factors, that could be affected and other mechanisms operating in the pathogenicity of Huntington disorder.

SLO 6. Describe how repeated DNA sequences and homologous recombination contribute to the appearance of interstitial deletion syndromes.
Microdeletion syndromes
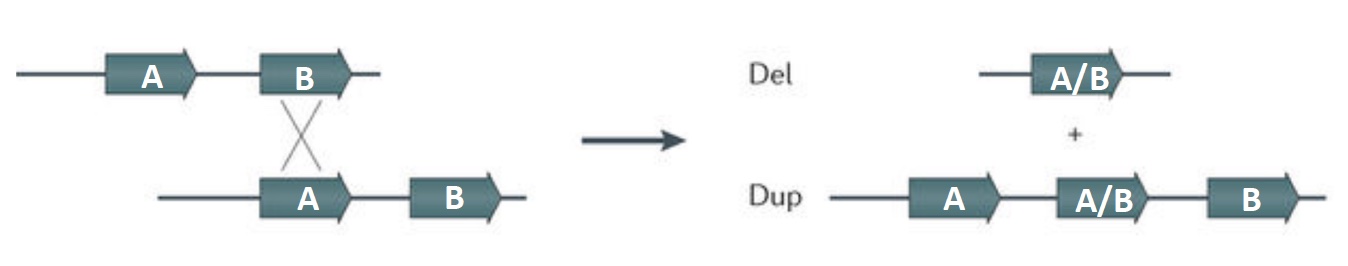
The introduction of double strand breaks (DSB) are part of the normal process of meiotic recombination during gamete development. The homologous recombination repair system is used in producing the meiotic recombinants. Homologous chromosomes may align out of register in areas where there are blocks of repeated sequences. If a crossover occurs when the wrong blocks pair, one of the resulting recombinant chromosomes will have a deletion between the repeats, and the other chromosome will carry a duplication. As this process is occurring in gametogenesis, the result will be a germline mutation affecting the next generation.
Small deletions (microdeletions) may be terminal (end of chromosome is deleted) or interstitial (internal region of chromosome is deleted). Frequently the deleted area is in between segmental duplications, which are multiple copies of tandemly repeated sequences at the breakpoints for the deleted region. This process is considered a nonallelic homologous recombination because it involves recombination between different genetic areas that may be similar but nonhomologous, as illustrated in the generic figure below.
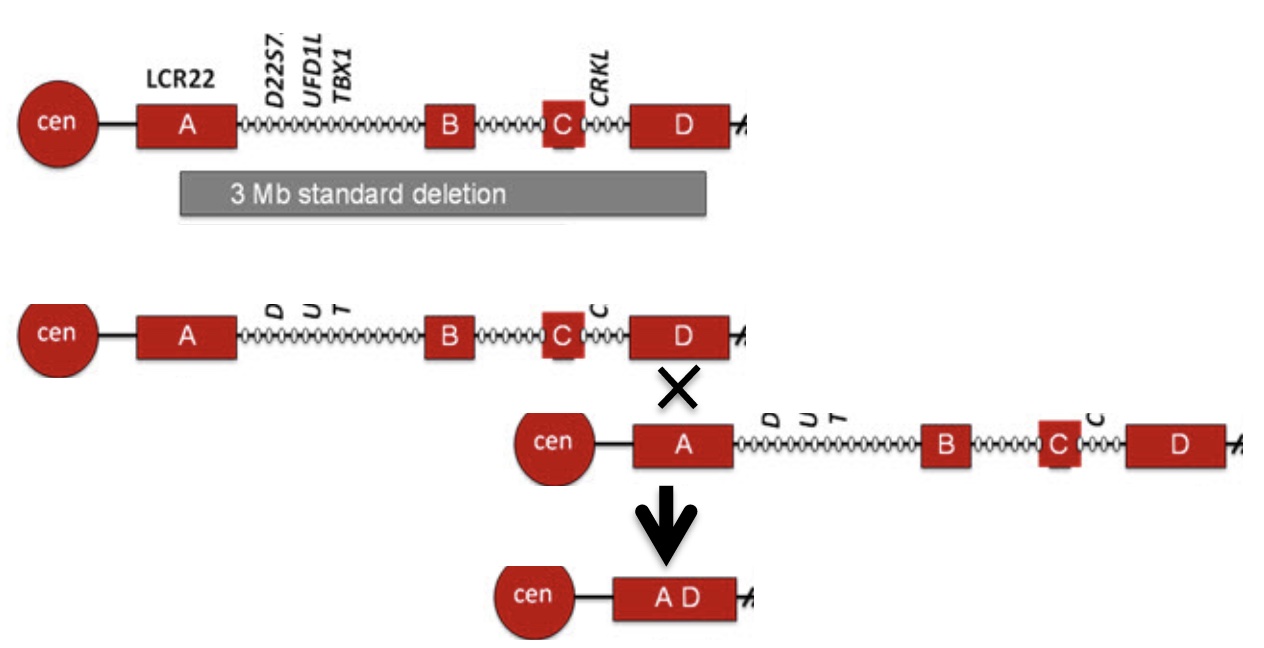
To relate these concepts to the most common microdeletion syndrome, the segmental duplications involved in 22q11.2DS (DiGeorge syndrome) are illustrated below, showing only the final deletion product after the nonhomologous recombination. Regions A and D have high sequence identity but are nonhomologous.
Microdeletion syndromes are designated by the chromosomal region affected or by their eponymous name. The “q” followed by a number indicates the chromosomal region affected on the long arm, and the “p” refers to the short arm. Some of the disorders that have been studied more extensively are listed below, but epidemiological studies have included several other microdeletion syndromes not described here. Because these disorders occur at a greater frequency than one might expect, the estimates reported by the National Organization for Rare Disorders (NORD) for the incidences in live births, however inaccurate because of misdiagnoses and underreporting, are included below to highlight the more common microdeletion disorders and those represented in USMLE First Aid book or in lecture.
- 22q11.2: DiGeorge syndrome (Velocardiofacial syndrome)
- o Common interstitial microdeletion syndrome presenting with many different congenital anomalies
- o Interstitial microdeletion syndrome with incidence of ~1/3000-1/6000 live births
- 7q11.23: Williams-Beuren syndrome (Williams syndrome)
- o Interstitial microdeletion syndrome with incidence of ~1/7500 live births
- o Associated with supravalvular aortic stenosis and renal artery stenosis
- o 7q micro deletion involves 27 genes, including elastin gene, may be included in diagnostic tests for haploinsufficiency
- o Sometimes refered to as “Happy syndrome” because of the highly social personality; some have hypothesized a connection between syndrome and folklore tales of people with elfin qualities and magical powers
- o Armellino Center of Excellence established at University of Pennsylvania in June 2022, launched by Dr. Jocelyn Krebs (WWAMI-AK), former president of the Board of Trustees for the Williams Syndrome Association and Williams syndrome researcher.
- 15q11.2-q13: Prader Willi (PWS) and Angelman (AS) syndromes
- o Interstitial microdeletion syndrome with incidence of ~1/10,000-30,000 for PWS and ~1/12,000-20,000 for AS live births
- o Low copy tandemly repeated sequences are present at the common breakpoints (BP1, BP2, BP3) flanking the deletion regions (rare deletions use additional deletion breakpoints)
- o Presentation of disorder from interstitial deletions is affected by parent of origin imprinting discussed in other sessions of the WWAMI foundations curriculum
- 5p deletion: Cri du chat syndrome
- o Incidence of ~ 1/15,000 – 50,000 live births
- o Terminal or interstitial deletions with many different break points
- o No known correlation between deletion size and severity of disorder
- 4p deletion Wolf-Hirschhorn syndrome (terminal microdeletion)
- o Incidence of ~ 1/50,000 live births
- o Deletion of most terminal portion of 4p (about ½ of short arm)