
9 Cystic Fibrosis and Mutation-Specific Therapies
MOLB: Cystic Fibrosis and Mutation-Specific Therapies
This chapter addresses the SLOs below, however the chapter is not divided into SLO sections.
Session Level Objectives (SLOs): after completing the session, students will be able to:
SLO 1. Explain the molecular and cellular basis of cystic fibrosis.
SLO 2. Explain how a single mutation can cause different manifestations in a variety of tissues.
SLO 3. Explain how CF treatments can ameliorate symptoms and predict difficulties implementing them effectively in patients.
SLO 4. Describe the advantages and limitations of mutation-specific molecular therapies.
CF is the most common life-shortening single-gene disorder in the U.S. and Northern Europe, affecting ~30,000 persons in U.S. alone. It is an autosomal recessive disorder. In the US, 1 in 28 white people are carriers; the incidence is much smaller in other ethnic groups (in people of African and Mediterranean origin, sickle cell anemia and G6PD deficiency are more common than CF; those diseases will also be covered in FMR). The median life expectancy has gradually increased from ~5 years before the 1950’s to >40 years today in the U.S. with the development of better treatments. However, a 2017 study in the Annals of Internal Medicine reported that life expectancy with CF in Canada was 10 years longer than in the U.S., and the gap has been widening!
In infants, CF commonly presents first as failure to thrive. This is due to blockage of the pancreatic duct, leading to incomplete digestion of food. Malabsorption and caloric needs persist throughout a CF patient’s life, but the most severe morbidities and mortality of CF in adults are due to the pulmonary diseases that develop.
Newborns are screened for CF in all states, initially using a blood test for trypsinogen (a pancreatic enzyme) that has a 90% false positive rate. Many states do routine DNA testing for the most common mutations. Diagnosis is usually confirmed with a sweat chloride test (CF patients have more salty sweat). Since treatments are now available that are specific to some of the mutations responsible for the disorder, a DNA test to determine the gene defect is always indicated if this test is positive.
CF is caused by mutations in both alleles of the CFTR gene on chromosome 7, resulting in a lack of functional Cystic Fibrosis Transmembrane conductance Regulator protein (CFTR), an epithelial cell plasma membrane chloride channel that also regulates other ion channels in the same cell. Ion channels allow ions to flow across the membrane in the direction that is down a concentration or potential gradient.
The severity of CF symptoms varies with the type of mutation; the most common mutation results in misfolding and degradation; it causes complete absence of CFTR from the plasma membrane. Therefore, homozygotes for this mutation have severe symptoms. Other mutations that still retain some CFTR functionality lead to milder forms of CF.
A life-threatening clinical manifestation of CF is buildup of thick, viscous mucus in the airways (CF is sometimes called mucoviscidosis in Europe), causing partial or total occlusion and allowing bacteria to colonize. Lack of chloride secretion, due to absent or defective CFTR, results in retention of Cl– and Na+ ions, and therefore retention of water in the lung epithelial cells. This causes the airway surface fluid layer to dry up. The viscous mucus can no longer be effectively cleared by the beating cilia, and this facilitates colonization of the airway by opportunistic biofilm-forming pathogens, such as Pseudomonas aeruginosa, Staphylococcus aureus and Burkholderia cepacia. Even though the bacteria can usually be controlled with antibiotics, they are never completely eliminated, and recurrences of lung infections are frequent. The bacteria induce an inflammatory immune response, primarily of neutrophils, which produce reactive oxygen species to kill the bacteria. Consequently, collateral damage to the lung tissue accumulates over the years, resulting in inflexible scar tissue that diminishes breathing capacity. Ultimately, due to the inflexibility and mucus accumulation, the lung capacity becomes so diminished that a lung transplant becomes the only option. Donor lungs are in short supply, and the operation is risky (it is often a combined heart-lung transplant). It can result in dramatic and immediate improvement in breathing, but can also lead to rejection of the donor lung or to additional opportunistic infections because of the immunosuppressant drugs that are given to prevent rejection.
Similar osmotic effects cause the secretions of the pancreas and biliary system to become thick and viscous. As a result, digestive enzymes (proteases, lipase, amylase) and bicarbonate produced in the pancreas do not reach the duodenum. This results in maldigestion and nutrients being lost in the stool. In the liver, the same process blocks the bile duct and leads to a deficiency of bile salts, which are needed to solubilize fats in the lumen of the small bowel. This further worsens fat absorption, leading to fat in the stool and deficiency of fat soluble vitamins. Malnutrition (“failure to thrive” in infants) is a major problem in CF. Keeping CF patients properly nourished is important, but difficult. Besides having problems digesting and absorbing their calories, their caloric need is higher due to difficulty breathing. CF also results in decreased appetite which can lead to fewer calories taken in. When their nutritional needs are properly managed, CF patients do better in terms of lung function and overall health. In adults, accumulated damage to the pancreas can lead to lack of insulin or delayed insulin secretion from the pancreas; this results in CF-related diabetes.
CF affects other organs with epithelial cells, including the skin (salty sweat and reduced sweat volume) and reproductive organs (males are almost always infertile due to absence of the vas; females can also be infertile, but there are cases of well-treated women with CF successfully having children).
Standard CF treatments are designed to ameliorate the symptoms, and have led to dramatic increases in life expectancy over the past decades. They don’t cure the disorder, so it is important to consistently apply them to prevent long-term deterioration. Adherence can be a challenge for children and adolescents, because the daily treatments do not provide visible short-term results. These treatments include: 1) Chest compressions to loosen mucus; 2) Inhaling hypertonic saline (using a nebulizer) to rehydrate mucus; Pulmozyme (DNase) can be added to reduce the viscosity of mucus by cutting up DNA released from lysed cells, and bronchodilators (e.g. albuterol, a β2 adrenergic receptor agonist) are used to relax airway smooth muscle. 3) Inhaled and oral antibiotics (e.g. Tobramycin or azithromycin, which inhibit bacterial protein synthesis). 4) Pills containing pancreatic digestive enzymes (amylase, lipase, proteases) taken with meals. 5) High calorie diet.
CFTR Structure and Function:
CFTR contains a bundle of transmembrane helices that form a chloride-permeable channel and three cytoplasmic domains: two nucleotide binding domains (NBD), and a regulatory (R) domain. Opening the channel requires phosphorylation of the R domain at multiple sites by cAMP-dependent protein kinase (PKA), as well as ATP binding and hydrolysis by the NBDs. CFTR is related to the ATP-driven pump P-glycoprotein, which drives many chemotherapy drugs out of cancer cells and leads to multi-drug resistance when overexpressed. However, CFTR is a channel, not a pump, which means it can only allow ions to flow down an electrochemical gradient.
Although the number of CFTR molecules in a cell membrane is small, they have a large effect on osmoregulation (it doesn’t take a lot of open channels to let a lot of ions flow). CFTR also regulates another chloride channel and a sodium channel.
The most common mutation that causes CF is due to a deletion of 3 bases. It is called ΔF508, because the mutant protein is missing a Phe residue (F), amino acid #508 out of the 1480 in the sequence. It is in the NBD1 domain, but not involved in ATP binding.
CFTR Gene:
The CFTR gene contains 24 exons spanning almost 200,000 base pairs on chromosome 7. Since less than 4,500 bases are needed to code for the amino acids in the protein, most of the sequence is in the introns. More than 2,000 mutations have been discovered in the CFTR gene, although only about 200 of these have been clearly shown to cause CF. As seen in the diagram below, they are scattered all across the gene.
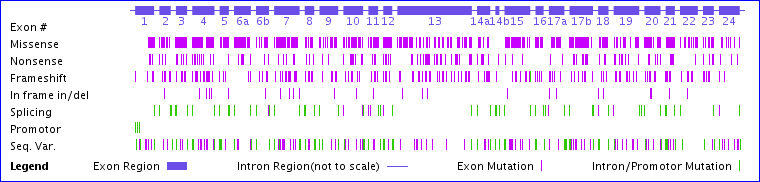
Missense mutations change a codon to specify a different amino acid, usually due to a single base change.
Nonsense mutations change a codon to a stop codon.
Frameshift mutations result when 1 or 2 bases are inserted or deleted in an exon (or any number not a multiple of 3). The remaining sequence downstream will code for the wrong amino acids and usually result in a truncated protein due to a stop codon in the other reading frame.
In frame insertions or deletions will result in one or more amino acids added or missing, but the rest of the sequence will be correct. The ΔF508 mutation is one of these.
Splicing mutations generally occur due to base changes at the exon/intron boundary and can result in an intron being retained in the mRNA and translated (usually leading to an incorrect and truncated sequence) or result in an exon being skipped. This may also result in a frameshift in the next exon.
There are many possible ways a mutation can lead to CF, and there are examples for almost all of them. A mutation in the promoter may result in inadequate transcription. A frameshift or splice junction mutation can result in a protein that bears little resemblance to CFTR. Such dysfunctional polypeptides are typically rapidly degraded to prevent protein aggregation. CFTR with the ΔF508 mutation, even though it has only one missing residue out of 1480, causes the protein to misfold and then be degraded before it ever gets to the plasma membrane. Other mutant CFTR proteins that reach the plasma membrane may be dysfunctional, either because they don’t open when they are supposed to (dysregulation) or don’t conduct enough chloride ions when they are open. Some mutations result in partial function due to reduced synthesis, reduced proper splicing (activation of alternate splicing), or more rapid degradation (turnover).
The geographical distribution of CFTR mutations varies widely. In people of Northern European descent, 90% of the mutant alleles are ΔF508; in Ashkenazi Jews, 48% are W1282X. These are called “founder effects”. In native populations of Sub-Saharan Africa and Asia, CF is relatively rare.
An understanding of the effects of a mutation can lead to drug therapies targeted to the effect of a particular mutation. Only 5% of normal CFTR function can help prevent lung and pancreatic disease symptoms, so even minor improvements in expression or function can have a big impact. Three examples that have led to new CF therapies are given below:
G551D:
This mutation has an Asp substituted for the Gly normally at position 551. It is found in 3-4% of CF patients, and results in a channel that does not open properly even though it is present in the membrane. The Cystic Fibrosis Foundation funded a project that first yielded a compound called a “potentiator”, because it increases chloride flow through G551D (as well as through normal CFTR and some other mutant forms). When it entered clinical trials it was named ivacaftor. A phase III study led from UW in 2011 showed significant improvements in G551D patients taking this drug in addition to standard therapy. Note that most of these patients have ΔF508 in their other CFTR allele, but only one of the genes needs to produce functional protein. When the FDA approved this as a new drug in 2012, it was given the brand name Kalydeco. In 2017, the FDA expanded the approval to treatment of 33 rare mutations, which doubled the number of patients for which Kalydeco was indicated.
ΔF508:
The most common mutation. Key observations were: 1) Individuals homozygous for the ΔF508 mutation transcribe and translate the gene, but have no CFTR in their plasma membranes. 2) When researchers expressed a human CFTR gene with the ΔF508 mutation in insect cells grown at 28C, the protein was found in the plasma membrane of these cells and it functioned as a chloride channel. These observations led to the hypothesis that the ΔF508 CFTR is absent in the plasma membranes of CF patients because it misfolds at 37C, but is stable enough to fold at 28C. Misfolded proteins are exported from the ER and degraded via the ubiquitin-proteasome system. This led to a search for small molecules (called “correctors”) that could help stabilize ΔF508 CFTR a little and get it to fold properly. The first compound that did this successfully was lumacaftor. It did not help patients by itself, but in 2014 a Phase III trial (again led by UW) in combination with ivacaftor reported improvement in most ΔF508 homozygotes. The FDA approved this combination (called Orkambi) in 2015. It only improves lung function (FEV1, the volume of air that can be exhaled in 1 min) by a few %, but every bit helps. Second generation correctors tezacaftor and elexacaftor in triple combinations with ivacaftor (called Trikafta) increased FEV1 by 14% in clinical trials. Trikafta was approved in 2021 for use in patients with one or two ΔF508 alleles.
G542X, W1282X:
Nonsense mutations are present in about 5-8% of CF patients. Ataluren was developed to cause read-through of stop codons due to nonsense mutations, of which G542X and W1282X are the most common in CF. A Phase III trial reported in 2012 showed modest improvement and slowed disease progression in CF patients with nonsense mutations. The drug was licensed in Europe, but is not approved in the US. Another Phase III trial in 2017 showed no significant improvement in FEV1. The FDA also rejected it for use in muscular dystrophy that year. What problems might be caused by a drug that causes read-through?
Gene therapy for CF has been attempted by various means without success since the 1990’s. One major obstacle has been targeting it to the airway stem cells that constantly regenerate the airway epithelia. In 2020 researchers demonstrated that they could take a patient’s blood cells, reprogram them to generate induced pluripotent stem cells (iPSCs) and then coax them into becoming basal airway stem cells. This opens the possibility of correcting a CF mutation in these cells and returning them to the patient.
A few final comments:
Every genetic disorder is “rare”, except in certain in-bred populations (e.g. the Faroe islands have the highest incidence for CFTR ΔF508); most disorders have incidences of 1 in 10,000 or less.
However, there are thousands of known genetic disorders, so the possibility that a patient has one of them should not be discounted.
Mutations occur almost everywhere and are found in everyone. We all have mutations in a number of important genes. Most are recessive, but we are all carriers of genetic disorders.
Missense mutations are the most common; they can be benign or severe depending on the substitution.
Nonsense mutations and frameshifts almost always result in translation of dysfunctional protein that could misfold and aggregate. Cells have mechanisms for degrading mRNAs with premature stop codons and degrading misfolded proteins to mitigate these effects.
Disease-causing mutations may be in introns or in promoter and enhancer regions thousands of bases from the coding sequences. Promoter and splicing mutations often result in reduced expression of normal protein, rather than complete absence.
If you sequence a gene (or a whole genome) and find variations from the “normal” DNA sequence of a gene, they may be harmless. Unless they have been found in other patients and characterized, you can’t assume they cause a disorder.
Although methods have been developed to replace or repair defective genes in the laboratory, translating that to a cure can be extraordinarily difficult. Think about what cells you would need to fix in a CF patient and how to get the DNA into them.
Additional information about CF can be found at Dynamed (under “Top Resources” on the UW Health Sciences Library website), and at the Cystic Fibrosis Foundation.
Feedback: