
4 Protein Structure and Function
Session Level Objectives (SLOs): after completing the session, students will be able to:
SLO1. Summarize the elements of protein secondary, tertiary, and quaternary structure.
SLO2. Describe the roles of hydrophilic vs. hydrophobic aminoacyl residues in protein folding.
SLO4. Describe the concept of a dissociation constant, Kd, for a general ligand-receptor pair.
SLO7. Describe how carbon monoxide affects the affinity of hemoglobin for oxygen.
SLO8. Explain a biochemical reason for how a fetus concentrates oxygen from its mother.
SLO 1. Summarize the elements of protein secondary, tertiary, and quaternary structure.
If you want to understand function, study structure. — F. Crick
As a nascent polypeptide emerges from the ribosome, it must fold into a specific, functional, three- dimensional structure. The functional “native” fold of a protein is determined by the linear sequence of amino acids in the polypeptide.
We think about folding as a hierarchical process:
- A polypeptide’s primary structure its linear sequence of amino acids. This sequence, as you have just seen, is specified by the sequence of codons in an mRNA template.
- Secondary structure elements are “folding motifs” that form through local interactions between residues within the polypeptide chain (H-bonding, salt bridges, van der Waals interactions, etc.). The most common and important secondary structure motifs are the α–helix (Fig. 1) and the β–sheet (Fig. 2).
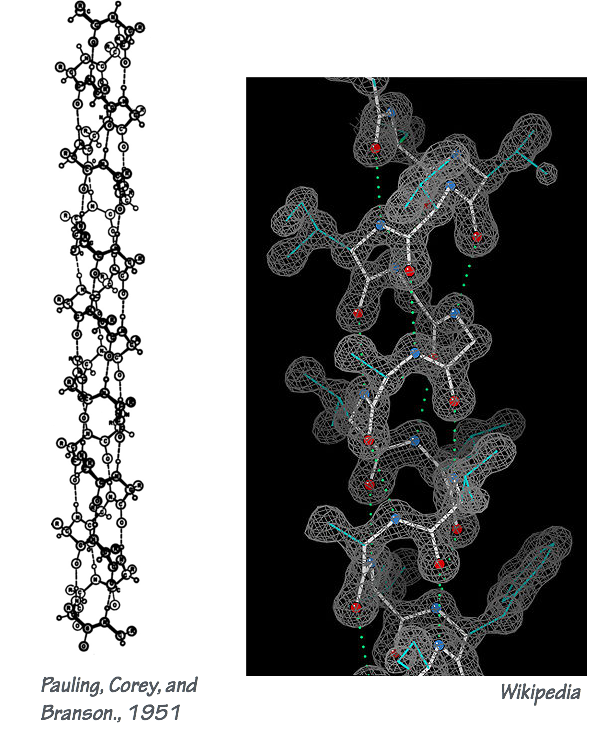
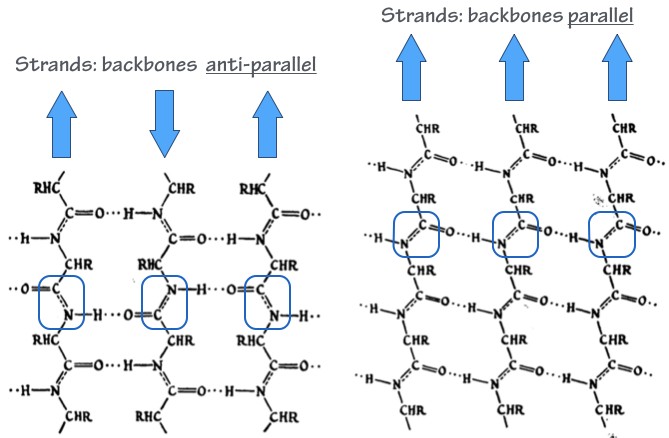
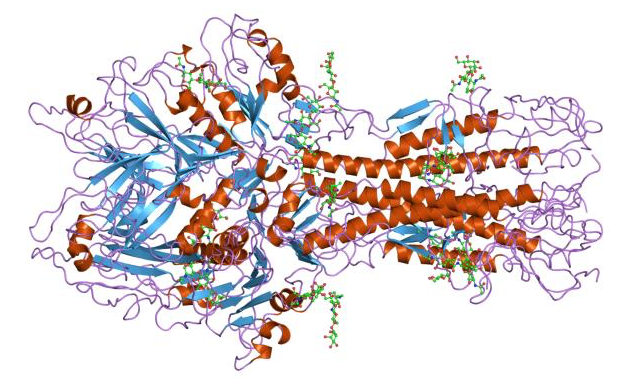
- Tertiary structure describes the overall arrangement of a polypeptide’s secondary structure elements. This is the overall 3-dimensional fold of the polypeptide. Many proteins contain mainly α-helix or β-sheet folds. Others use both kinds of folds; an example is shown in Fig. 3.
- Many, many proteins operate as larger complexes. The assembly of more than one polypeptide into a protein complex is the quaternary structure. This can mean as few as two small polypeptides, or an assemblage as big as the nuclear pore or — even bigger — silk or human hair.
Proteins come in a dazzling variety of shapes, sizes, abundances, and tissue distributions.
Nearly without exception, all proteins are functionally similar: what proteins do is recognize specific chemical entities, and bind — stick — to them.
That’s it. That’s almost (though not quite) the whole deal. Proteins stick to specific things. Antibodies stick to antigens to mediate immune responses during infection. Cell adhesion molecules allow cells to stick to each other and to extracellular matrix proteins. Extracellular matrix proteins stick to each other, and to cells. Transcription factors recognize and stick to specific enhancer sequences in our genes. Odorant receptors stick to specific volatile molecules (from perfume to putrescine). Hormone receptors stick to insulin, estrogen, or other hormones.
Enzymes are also most simply understood through their ability to stick to things. They stick to their substrates, and they stick to transition states more tightly, favoring the formation of those otherwise unfavored states, and accelerating reaction rates. Enzymes often bind products less tightly, allowing their dissociation (release) from the enzyme.
Within membranes, ion channels, transporters, and pumps are again sticking to substrates and using a series of binding steps to move things from one side of a membrane to the other.
Molecular motors like myosin do the same thing again. Myosin sticks and un-sticks to the actin thin filament. The order of myosin-actin sticking-unsticking is coupled to myosin sticking (binding) to ATP, to the ATP hydrolysis transition state, and finally to the release of ADP and Pi. Here, coupling of two binding cycles allows energy derived from ATP hydrolysis to make myosin’s binding and un-binding to actin directional — and that is the power stroke that makes our muscles contract.
With a general quantitative description of a protein’s binding characteristics we can understand an enormous amount about what a protein does, how it does it, and how it can fail in its functions, leading to pathology. The same concepts, as we will see in the next session, allow us to think about how drugs interact with their molecular targets.
After all, most drugs are just chemical entities that particular proteins recognize, and stick to.
SLO 2. Describe the roles of hydrophilic vs. hydrophobic aminoacyl residues in protein folding.
The principles that control protein folding are the exactly same ones that we have already seen with RNA and DNA: The hydrophobic effect, charge interaction and repulsion, Van der Waaals contacts, etc.
- Water molecules form many hydrogen bonds with one another, and they have high entropy (they can diffuse freely, translate, and rotate).
- Hydrophobic (greasy) amino acid side chains are surfaces where water cannot hydrogen bond (this is an enthalpic penalty). Near these surfaces the water has reduced entropy, as well. Because water “hates” hydrophobic side chains, these chains “want” to be shielded from the aqueous solvent. Thus, hydrophobic amino acid residues tend to be buried within folded portions of the protein.
- Hydrophilic (polar or charged) amino acid side chains can form energetically favorable hydrogen bonds with water. They are often exposed to the aqueous solvent. If they cannot interact with the solvent (if they are buried), they generally interact with other portions of the polypeptide through hydrogen bonds or salt bridges.
- Additional inter-chain interactions that contribute to protein stability include van der Waals contacts, aromatic stacking interactions (analogous to the base stacking that we saw with DNA and RNA), and electrostatic repulsion between similarly charged (-/- or +/+) groups on the polypeptide.
Figure 4.

SLO 3. Explain the importance of correct protein folding, chaperone proteins, and how misfolding can lead to pathology.
Protein folding, chaperone proteins, and how misfolding can lead to pathology
Mutations that cause protein sequence changes, or errors in transcription or translation, can change the balance of forces that we have just described, causing misfolding of a protein loss of its function. Other mutations may still allow a protein to fold more or less correctly, but change the protein’s activity. For example, some mutations result in ion channels that open more easily than they otherwise would, leading to neurological disorders.
Both within our cells, and in extracellular spaces (cartilage, blood, cerebrospinal fluid, etc.), proteins are present at extremely high overall concentrations. This dense proximity means that proteins will touch other proteins both during and after folding, with enormous potential for inappropriate interactions that can lead to non-specific aggregation.
You’re probably familiar with one protein aggregation process: making Jell-O™. We start with a clear aqueous solution of soluble proteins at high concentration. We then heat the solution so that the proteins unfold. That is, they are denatured. As the unfolded proteins cool, they aggregate into a single disordered gel.
In cells, protein aggregates are major sources of cytotoxicity, and — as we will see — they contribute to pathologies ranging from Alzheimer’s disease to type II diabetes.
Mutations can cause proteins to misfold at elevated rates, but even non-mutant proteins sometimes misfold, especially in the presence of stresses such as heat or oxidation. To mitigate inappropriate contact between un-folded or partially-folded proteins, cells use special proteins called chaperones. There are many different chaperones. Some passively shield proteins from inappropriate contacts.
Others use energy from ATP hydrolysis to mechanically pull apart proteins that have formed inappropriate contacts, giving the proteins a “second chance” to fold correctly.
When a protein cannot fold correctly even with the assistance of chaperones, the cell may recognize the misfolded polypeptide as hopeless, and mark it for destruction. This cellular surveillance process operates in almost every cell and is called protein quality control. The quality control system has at least two branches: the ubiquitin–proteasome system, and the autophagy–lysosome system. We’ll discuss these systems later in the block.
Common post-translational modifications and specific modifications that occur predominantly on proteins within the cytoplasm or in extra- cytoplasmic environments.
Once synthesized, most polypeptides undergo covalent post–translational modifications. These fall into several different categories. The following list is not comprehensive! It’s illustrative, showing some important examples.
-
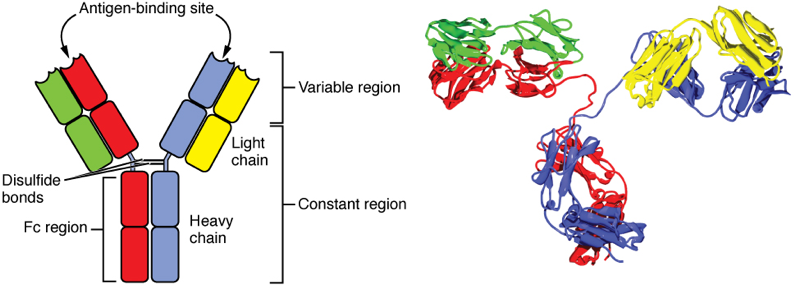
Fig. 5. An antibody (IgG) molecule. Each IgG is a heterotetramer containing four polypeptides: two identical light chains and two identical heavy chains. IgG is an entirely β-sheet protein. The quaternary structure of the complex is stabilized by non-covalent interactions between the chains, and also by disulfide bonds that covalently cross-link the two heavy chains together. Proteolysis. Many proteins are precisely clipped before they are fully functional. For example, many digestive enzymes are made as inactive proenzymes — a form safe for transport through sensitive cellular compartments. Upon secretion into the digestive tract an inhibitory portion of the polypeptide is clipped off, and the enzyme is activated.
- Disulfide bonding. The terminal sulfhydryl group on the amino acid cysteine can be oxidized to form a cysteine–cysteine disulfide bond.
- Disulfide bonds are most often used to form mechanically stabilizing cross-links within a polypeptide chain or to cross-link two chains together in a protein complex.
- In general, the cytoplasm and nucleus of a cell have a chemically reducing potential, while the extracellular environment has a relatively oxidizing potential. What this means: we very seldom see proteins with disulfide bonds in the cytoplasm or nucleus, but lots of secreted proteins such as antibodies (Fig. 5), and many cell-surface proteins, have disulfide bonds.
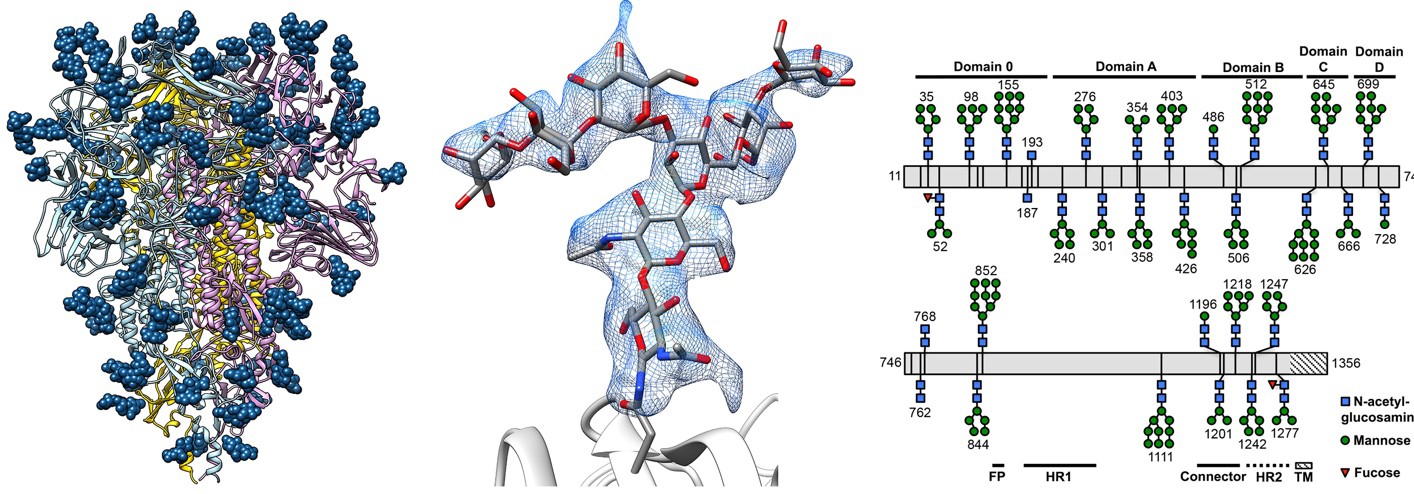
Fig. 6. The glycan “shield” of a Coronavirus spike protein. Like the Influenza HA protein shown in Fig. 3, the Spike protein is used by Coronaviruses to gain entry into host cells during infection. The spike consists of a protein homotrimer, anchored to the viral envelope (membrane) at the base. Attached to each monomer are over twenty complex carbohydrate molecules (blue). Each sphere represents a hexose. The structure of the carbohydrates, ascertained using mass spectrometry, is shown in schematic form on the right.The carbohydrates both stabilize the spike protein and shield it from proteases, antibodies, and other host defenses.Source: D. Veesler, UW Biochemistry. Nat Struct Mol Biol. (2016) 23(10):899-905
- Glycosylation. Sugars are attached to most, but not all, secreted and cell surface proteins. Usually these are short-chain, branched carbohydrates. These sugars are used in cell–cell recognition and in cell signaling processes, and they can stabilize and protect proteins that are exposed to harsh extracellular environments. On the down side, viruses such as HIV and SARS use glycosylation to shield their surface proteins from recognition and attack by our immune systems (Fig. 6). Very few cytoplasmic or nuclear proteins are glycosylated (though the ones that are may be of great importance).
- Phosphorylation. The covalent transfer of phosphate groups from ATP to polypeptides (protein phosphorylation) is a critical regulatory mechanism that controls almost every aspect of cell physiology.
- The phosphotransfer reaction is mediated by protein kinase enzymes. The recipient is always an amino acid residue with a terminal hydroxyl group on its side chain: serine, threonine, or tyrosine. The product is a phosphoester.
- Phosphorylation is reversible. Hydrolysis of the phosphoester removes the phospphoryl group from the protein. Dephosphorylation is catalyzed by protein phosphatase enzymes.
- Several hundred kinases and phosphatases are encoded in the human genome.
- Protein kinases and phosphatases were discovered here in the UW School of Medicine, by Professors Ed Krebs and Eddie Fischer. For their discoveries, these lifelong friends and collaborators shared the Nobel Prize.
Different proteins have different functions — and the proteins must carry out their functions in different locations. Inside a cell, for example, some proteins operate in the cytoplasm, some in the nucleus, some within organelles such as mitochondria, and some within the plane of the plasma membrane. Many proteins are secreted from cells. Examples include the collagen that holds our tissues together (Fig. 7), antibodies and other proteins in blood and serum, digestive enzymes in the gut, and polypeptide hormones such as insulin.
Each protein must have a way to get to its site of action. Protein targeting typically involves a specific amino acid sequence within the polypeptide that serves as a “molecular zip code” used to direct the protein to its destination.
For example, the RNA polymerase II complex consists of several polypeptides. It is synthesized, folded, and assembled into a complex in the cytoplasm, but it has a “nuclear localization sequence” that directs the folded complex through the nuclear pore and into the nucleus, where it will transcribe mRNA molecules. As we’ll see, some mutations cause proteins to go to incorrect locations, resulting in disease.
SLO 4. Describe the concept of a dissociation constant, Kd, for a general ligand-receptor pair.
O2 is a ligand, and we can say that HbA is a receptor that binds it. As the partial pressure of oxygen, pO2, increases, the fractional saturation of HbA increases (Fig. 8).
The first thing to notice is this is an ensemble measurement of many molecules of HbA. We note that there is a concentration of O2 where 50% the receptor (HbA) is 50% saturated by its ligand (O2).
- This concentration is the dissociation constant (KD), and it indicates how tightly a ligand binds its receptor. For this reason, KD is also called the affinity constant.
- KD has units of concentration. For a gas, we may use partial pressure (units: mm Hg, atmospheres, Pa, pounds per inch2 (p.s.i.), etc.). For solutes in liquid we will more often use concentration per volume (M, g/L, etc.).
- We say that an interaction with a small KD (say, 10-9 M) is high–affinity. We say that an interaction with a large KD (say, 10-3 M) is low–affinity.
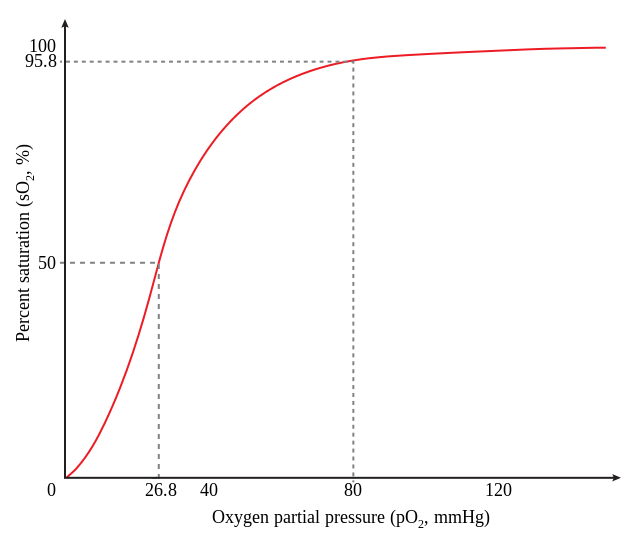
Fig. 8. Fractional saturation of HbA at different O2 partial pressures. Source: Wikimedia - For a simple ligand-receptor pair, L + R ⇌ LR, KD = ([L][R])/[LR] where [L], [R], and [LR] are the concentrations of the ligand, the receptor, and the complex, at equilibrium (when the concentrations of free and receptor-bound ligand are not changing).
- KD is an equilibrium constant, which reflects two rates: the rate at which a particular ligand sticks to the receptor (association), and the rate at which that ligand falls off (dissociation). For the simple case, L + R ⇌ LR, KD = koff/konwhere kon is the rate constant for association and koff is the rate constant for dissociation. kon has units of inverse concentration and time (M-1 s-1), and Koff has units of inverse time (s-1).
- Take the time to satisfy yourself that if the association rate constant increases, the receptor-ligand affinity increases. Conversely, if dissociation rate constant increases, the affinity decreases.
Now take another look at the shape of the saturation curve for HbA. What does the shape of the curve tell us?
Enzymes
Enzymes are highly selective catalysts.
- Enzymes bring together specific substrate molecules in specific geometries, and accelerate chemical reactions that convert the substrates into specific products.
- As catalysts, enzymes cannot change the thermodynamics — the overall free energy balance — of a reaction
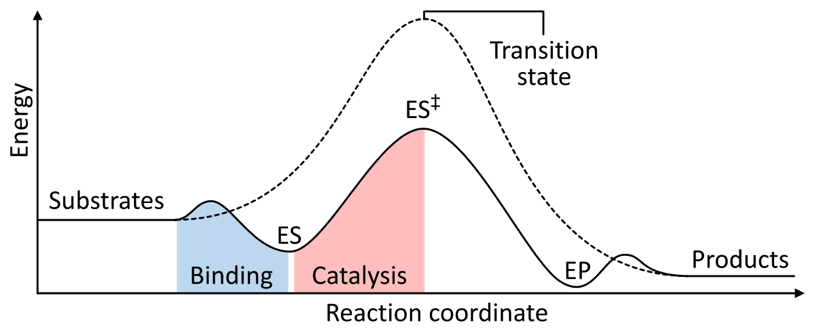
Fig. 9. Principle of enzyme-mediated catalysis. E, enzyme. S, substrate(s). P, Product(s). The enzyme does not alter the overall free energy balance of the reaction pathway. Rather, the enzyme lowers the activation energy for formation of the transition intermediate. Source: Wikimedia (Fig. 9).
- Instead, enzymes change the kinetics of the reaction. They do this by reducing activation energy barriers that would otherwise prevent the reaction from occurring at a biologically relevant rate.
- By bringing specific substrates together in the appropriate geometry, and by shielding intermediates from reactive non-substrate molecules, enzymes suppress the formation of off– pathway products.
- The vast majority of enzymes are proteins. (But, as we saw with the ribosome, a small critical subset of enzymes have catalytic centers made out of RNA.)
- Many enzymes use covalently or non-covalently bound prosthetic groups to control the electronic environment within their active sites, promoting catalysis. Prosthetic groups are sometimes, but not always, vitamins. Thus, vitamin deficiency often leads to compromised function in specific enzymes, leading to metabolic or structural pathologies.
- In summary, we can think of enzymes as receptors that bind ligands (substrates) in highly specific ways, to promote specific chemical reactions among those substrates.
As with ligand-receptor interactions, a small number of parameters provides a vivid description of an enzyme’s critical properties.
- V is the reaction rate at which substrate is converted to product, under some specified set of conditions (substrate concentration, temperature, pH, etc.).
- The Vmax is the maximum rate at which the enzyme can convert substrate to product. Vmax occurs when the substrate is at a sufficiently high concentration that its availability is not rate-limiting.
- The KM, or Michaelis–Menten constant, is the concentration of substrate at which the rate of product formation per molecule of enzyme is half-maximal. Thus, at a substrate concentration [S], where V = Vmax/2, KM = [S]
- KM is closely analogous to KD, because each refers to the concentration of ligand or substrate where 50% of the receptor or enzyme is occupied.
- kcat is the rate constant for the enzyme-catalyzed conversion of the enzyme-bound substrate to product: For the scheme in Fig. 8, kcat is the rate for ES ⟶ E+P. kcat is also called the enzyme’s turnover rate.
- The catalytic specificity of an enzyme for any given substrate can be defined as a ratio, kcat /KM
- By comparing kcat /KM ratios for one enzyme and various substrates, we can learn how selective an enzyme is. For this reason kcat /KM is often called the specificity constant.
- For example: the active site pockets of DNA polymerase enzymes have high affinity (small KM) for dNTP (DNA) nucleotides, but extremely low affinity (very large KM) for NTP (RNA) nucleotides. This is because DNA polymerase active sites are usually shaped so that the 2´-OH group of dNTPs does not fit within the pocket.
- Thus for DNA polymerases kcat /KM for dNTPs is relatively large, while kcat /KM for NTPs is very small. We therefore say that DNA polymerases are selective for dNTP substrates versus NTP substrates.
- As we’ll see in following sessions on Pharmacology, competitive enzyme inhibitors have high affinities for enzyme active sites (low KM), but small — or zero — kcat. Competitive inhibitors “clog” an enzyme’s active site, preventing legitimate substrates from binding.

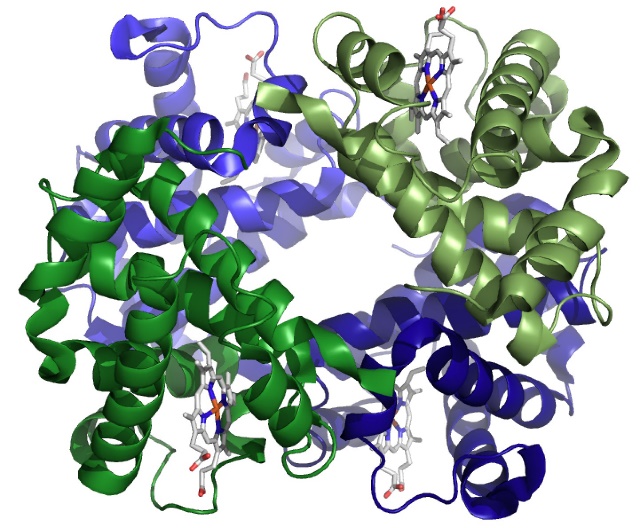
SLO 5. Describe the quaternary structure of hemoglobin and explain the function of the heme prosthetic group.
Hemoglobin (Hb) is a protein present at
enormously high concentration in the cytoplasm of red blood cells. The most important (but not only) thing hemoglobin sticks to is molecular oxygen (O2).
Structure of Hb
- Adult Hb (HbA) is a complex of four polypeptides: two α chains and two β chains (α2β2).
- Fetuses and infants have fetal HbF containing two γ (gamma) chains instead of β chains (α2γ2). The α, β, and γ chains are not identical, but they have very similar primary sequences and a nearly identical, all α-helical, tertiary fold.
- Terminology note: α -helices and β-sheets are not the same as the α and β chains of Hb.
- Separate genes encode mRNA templates for the various Hb chains.
- Each of the four chains cradles one heme molecule (Figs 11 and 12): a porphyrin ring that coordinates an ion of iron (Fe2+) at its cent
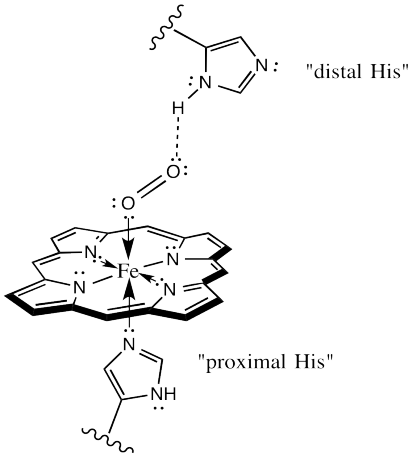
Fig. 12. Mechanism of O2 binding by Hb. The proximal His (histidine) is covalently bound to the heme iron. The distal His helps to coordinate the O2 molecule. er. The bound heme is an example of a prosthetic group — a non- amino acid bound to an enzyme and required for its activity. As we’ll soon see, vitamins often serve as prosthetic groups in enzymes.
- Within each subunit (or chain), amino acid side-chains and the bound heme operate together to bind one O2 molecule (Fig. 12). Thus, the binding capacity of a hemoglobin heterotetramer is 4 O2.
- It follows that a one Hb tetramer can be ¼, ½, ¾, or completely saturated with O2.
- With huge numbers of Hb molecules, O2 saturation of the population can be anywhere from 0 to nearly 100%.
SLO 6. Explain how allosteric cooperativity in hemoglobin enhances oxygen delivery to peripheral tissues, especially during exercise.
Function: what Hb needs to do
As erythrocytes pass through our lungs, their Hb binds O2. The erythrocytes flow with the blood to our peripheral tissues where they dump the O2. The tricky bit is that Hb needs to hold on to its precious cargo of O2 until it is in a part of the body that’s most in need of O2.
In other words, Hb needs to bias its O2 binding characteristics to accelerate dissociation in relatively hypoxic environments, rather than dumping O2 randomly. This, along with control of vasoconstriction, allows us to maintain a relatively shallow pO2 gradient from our lungs to our fingers, toes, and brain in the periphery.
The affinity of Hb for O2 is affected by temperature and pH and concentrations of CO2 and 2,3-DPG. During exercise there is a right shift in the Hb-O2 saturation curve because of increased CO2, acidity, DPG and temperature. Consequently, Hb unloads O2 more easily in the actively metabolizing tissues.
SLO 7. Describe how carbon monoxide affects the affinity of hemoglobin for oxygen.
SLO 8. Explain a biochemical reason for how a fetus concentrates oxygen from its mother.
The beta-globin chains of adult hemoglobin HbA have a greater affinity for 2,3-DPG than do the gamma-globin chains of fetal hemoglobin HbF. Because binding 2,3-DPG lowers the affinity of hemoglobin for O2, the lower affinity of HbF for DPG causes HbF to have a higher affinity for O2. Consequently, the presence of HbF in the fetus allows the fetus to concentrate O2 from the mother during pregnancy.
Feedback: