
10 Proteasome, Lysosome Function
Session Learning Objectives:
SLO1. Explain the concepts of protein quality control and surveillance.
SLO2. Delineate the two major pathways of protein turnover (1): the ubiquitin-proteasome pathway.
SLO3. Delineate the two major pathways of protein turnover (2): the lysosome/autophagosome pathway.
SLO4. Outline the genetic bases of major lysosomal storage diseases.
Overview of Protein Degradation
SLO1. Understand the concepts of protein quality control and surveillance.
As much as 30% of newly synthesized cellular proteins are discarded without being properly folded (i.e. misfolded, and/or unassembled), even though they may be synthesized normally without mutations of their genes or errors in the translation process. In addition, even if proteins are synthesized and folded accurately as functional proteins with normal tertiary structures, they often undergo damage over time due to various environmental stresses, such as heat, oxidation (i.e. from free radicals), and UV exposure. In healthy individuals, these impaired proteins with non-native or aberrant structures are rapidly degraded. In other words, the cell is fully equipped with a “surveillance system” to rapidly eliminate these defective proteins. The molecular chaperone system recognizes proteins with non-native structures, prevents them from irreversibly aggregating, and facilitates their proper refolding. The ubiquitin-proteasome system (UPS) is responsible for selective destruction of misfolded/unfolded and unassembled proteins which have failed to correctly refold by the chaperone system. This machinery prevents the accumulation of abnormal proteins and formation of toxic aggregates, as seen in various neurodegenerative diseases (covered in later sessions).
Other proteins need to be removed from the cell when conditions change (e.g. downregulation of receptors) or their task is completed (e.g., proteins involved in cell division). Most proteins are subject to continuous turnover (i.e. a constant process of protein synthesis and degradation). Some individual proteins may have half-lives of only a few minutes, while some last weeks or even years (proteins in tendons and eyes). The concentration of a protein is thus regulated at both the synthesis and the degradation level. Foreign proteins from bacteria or viruses must also degraded, and sometimes proteins must be metabolized as a source of energy or building blocks for other needed molecules. Whatever the purpose, the two major mechanisms by which cells degrade unwanted proteins, depending on their origin or cellular location, are the ubiquitin-proteasome system (UPS) and the lysosomal pathway.
Clinical significance of protein degradation
The UPS is capable of catalyzing rapid, timely protein degradation necessary for a multitude of biological processes, including the cell cycle and programmed cell death (apoptosis). In addition, the UPS plays a major role in the stress response and protein homeostasis.
Proteasome inhibitors have been in clinical use for many years in the treatment of multiple myeloma. They are thought to work by inhibiting the degradation of proteins required for apoptosis. Most cancer chemotherapies trigger apoptosis, and cancer cells can become resistant to these chemotherapies by rapidly degrading proteins in the apoptotic cascade and prolonging the life of the cell.
Proper immune system function relies on the generation of immunogenic peptides by degradation of foreign antigens and their display on the cell surface bound to MHC proteins. Both the proteasomal and lysosomal pathways are involved; this will be discussed more in the I&I block.
SLO2. Delineate the two major pathways of protein turnover (1): the ubiquitin-proteasome pathway.
The ubiquitylation process: tagging of proteins marked for degradation
Ubiquitin (Ub) is a highly conserved small protein, found in all eukaryotes, that acts as a degradation marker for a wide spectrum of cellular proteins. Functional analogs of ubiquitin have been described in some prokaryotes.
Ubiquitylation (also called ubiquitination) involves 3 enzymes: E1 activates the C-terminal end of Ub and transfers it to E2, and then one of many E3 (ubiquitin-protein ligases) transfers the Ub to a Lys sidechain of the target protein. A polyubiquitin chain can be formed using the same set of enzymes, by attaching another ubiquitin through its C-terminal end to a Lys residue within the ubiquitin previously associated with the target protein
A polyubiquitin chain functions as the main marker for proteolysis by the proteasome (a large, ATP-dependent, multi-subunit degradation complex).
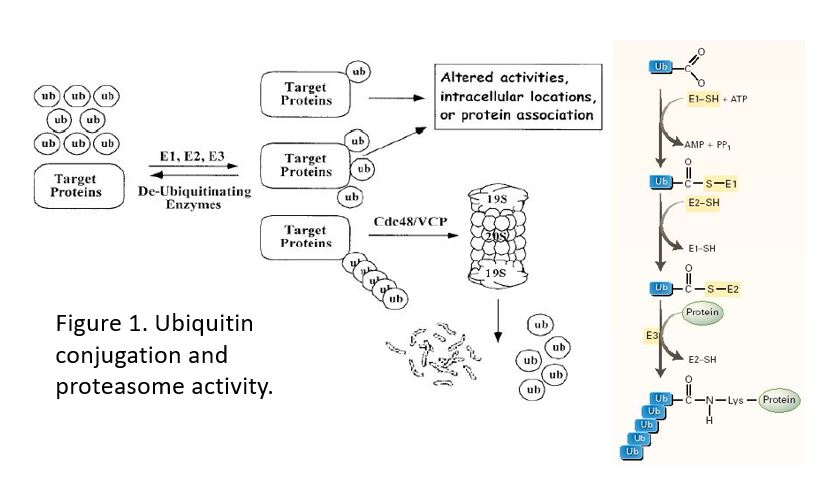
Figure 1. Addition of a single ubiquitin is used to target a protein to a specific location (e.g. the EGFR to lysosomes) or to modify the activity of a protein. Only a chain of 4 or more ubiquitins targets a protein to the proteasome.
Selectivity of protein degradation: Determined primarily at the stage of ligation to ubiquitin
There is a single E1 (activating) enzyme, whereas there are 30 E2s in multiple families and >600 E3 (ligase) enzymes needed to recognize the very diverse target proteins for degradation. Thus, E3 plays a critical role in the selection of the target protein for degradation, because each distinct E3 binds a protein substrate or set of substrates with a high degree of selectivity.
• Primary signal: E3 recognizes small primary sequence motifs on protein substrate
• Secondary signal: the poly-ubiquitin chain is recognized by the proteasome
• Ub4 is shortest chain that binds to the proteasome
• Cells also have deubiquitylation enzymes (DUBs) to facilitate removal of ubiquitin tags when a target protein enters the proteasome and is recycled (reused)
• DUBs also catalyze reversal of the mono-ubiquitylation reaction when protein is not to be degraded
Why is there a polymeric signal like poly-Ub for protein degradation?
1. Poly-Ub allows the proteasome to interact with vastly different protein substrates.
2. Balance between ubiquitylation/deubiquitylation processes may fine-tune the rate of degradation.
3. Ubiquitin tagging of proteins is not solely to target proteins for degradation. Mono-ubiquitylation is involved in other cellular events, such as: activation of kinases or transcription factors, DNA repair, endocytosis of membrane receptor proteins, and intracellular trafficking of proteins.
Proteasomes: Controlling protein degradation through compartmentalization
Protein degradation poses real hazards to a cell – therefore it is compartmentalized either in proteasomes or in degradative organelles such as lysosomes.
The proteasome is a large ATP-dependent protease complex, consisting of a central catalytic/core particle and two terminal regulatory caps attached to both ends. The “core” of the proteasome is a barrel-like cylindrical particle formed by the axial stacking of four rings, each of which is made up of seven structurally similar subunits. Subunits of each inner ring have the catalytically active residues– with three distinct protease specificities in the different subunits. These active sites face the interior of the cylinder, and substrates gain access to the active sites only after passing through a narrow opening at the center of the outer rings.
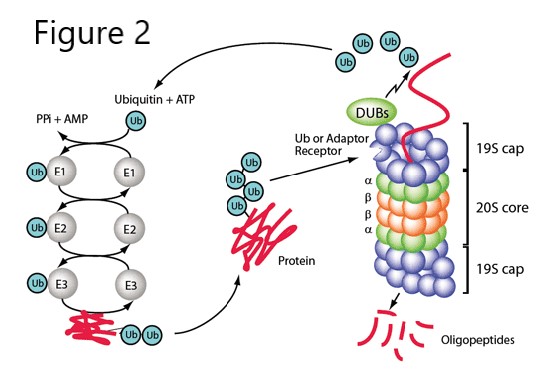
Figure 2. Ubiquitin conjugating enzymes and the proteasome.
The lid complex is involved in the recognition of target proteins, deubiquitylation, and interactions with various proteins, including E3s. As substrate proteins are threaded through the proteasome, they are cleaved into peptides 7-9 residues long. These are just the right size for binding to MHC class I receptors for display to the immune system on the cell surface. Peptides from degradation of both normal cellular proteins and foreign proteins are presented to cytotoxic T-cells in this way. It is up to the T-cells to recognize which are “self” and which are “non-self” (more on that in the I&I block).
Other functions of ubiquitin
The covalent attachment of Ub to a target protein can also serve as a localization signal, rather than a signal for destruction, by acting as a protein–protein interaction domain in endocytosis, membrane fusion and transcriptional control. For instance, mono-ubiquitylation stimulates endocytosis of cell surface receptors and progression through the endosomal sorting complex. Receptors that are not ubiquitylated are recycled back to the cell surface, but those that are ubiquitylated are more likely to be degraded instead. For example, increased stimulation of the epidermal growth factor receptor (EGFR) leads to downregulation by increased EGFR ubiquitylation and lysosomal degradation.
Ubiquitin’s involvement in the lysosomal pathway is also exemplified by a familial form of Parkinson disease (PD) that is caused by recessive mutations in PARK2, a gene encoding Parkin, an E3 ubiquitin ligase. Parkin is involved in tagging damaged mitochondria for degradation by lysosomes (mitophagy, described below).
SLO3. Delineate the two major pathways of protein turnover (2): the lysosome/autophagosome pathway.
The endocytic system, autophagy, and lysosomes
This organelle system targets both intracellular and extracellular substrates for destruction in the lysosome. Extracellular substrates are taken up through endocytosis (in clathrin-coated vesicles), pinocytosis, or phagocytosis. The endocytic or phagocytic vesicles containing these substrates are acidified by ATPases that pump protons into the lumen, and then the vesicles dock and fuse with lysosomes. The contents of the hybrid compartment are destroyed by lysosomal hydrolases (proteases, glycosidases, nucleases, etc., that function at low pH). Intracellular substrates, such as complexes or aggregates of proteins, are taken up by autophagosomes, vesicles that wrap around and enclose them. The autophagosome then fuses with a lysosome, allowing it to destroy the enclosed contents. Autophagy can occur in bulk, during starvation. This recycles protein and RNA building blocks for the cell. Autophagy can also function as a quality control mechanism that removes entire damaged organelles, such as mitochondria (“mitophagy”).
The lysosome is the terminal compartment in the endocytic pathway. It is also the terminal compartment of the autophagy pathway. The lysosome’s lumen (interior) is acidic (pH 4.5-5) and contains a variety of hydrolases that function at low pH and degrade macromolecules. The lysosome also serves as a storage depot for amino acids and metal ions.
Lysosomal hydrolases are synthesized at the rough ER where they are folded and glycosylated. They are then transported to the cis-Golgi where the carbohydrate is terminated by addition of mannose-6-phosphate (M6P). At the trans-Golgi, the M6P receptor is the adapter that picks the hydrolases and places them in a carrier vesicle. The carrier vesicle fuses with the late endosome, which is then delivered to the lysosome. The empty M6PR is then recycled to the trans-Golgi to pick up additional cargo.
Peptides generated by proteolysis in the lysosomes can also be displayed on the cell surface for recognition by the immune system, just like those produced by the proteasomes. In the phagolysosomes of phagocytic cells, peptides derived from endocytosis and digestion of bacteria or viruses are bound by MHC class II receptors, which are cycled back to the plasma membrane in vesicles with their cargo bound. Once the vesicles fuse with the plasma membrane, the peptides presented on the cell surface by the MHC II proteins are recognized by T cell receptors on helper T cells, and this helps activate an immune response to the pathogen. Again, you will learn more about this in I&I.
SLO4. Outline the genetic bases of major lysosomal storage diseases.
Lysosomal storage disorders
A variety of inherited lysosomal storage diseases are caused by deficiency of lysosomal hydrolases.
Examples include:
• Gaucher (glucocerebrosidase = β-glucosidase)
• Fabry (α-galactosidase)
• Tay-Sachs (hexosaminidase)
• Nieman-Pick (acid sphingomyelinase)
• Pompe (α-glucosidase).
Some of these diseases can be treated (not cured) through supplementation with the purified enzyme that is endocytosed and targeted by the M6P receptor. For example, Cerezyme (imiglucerase) is a recombinant β-glucocerebrosidase used to treat Gaucher disease. Cerdelga (eliglustat), a small molecule inhibitor of glucosylceramide synthase that reduces the need for lysosomal degradation is also used. Either treatment costs up to $300,000/yr. Enzyme replacement therapy with recombinant α-galactosidase for Fabry is similarly expensive. On the other hand, there is only palliative treatment available for the autosomal recessive disorder Tay-Sachs. Most babies born with the more severe, infantile form of Tay-Sachs, are homozygous for defective hexosaminidase alleles harboring a 4-base pair insertion in exon 11. GM2-ganglioside accumulates in cells, leading to a mental and physical deterioration that is usually fatal before the age of 5. Clinical trials of gene therapy are underway using injection of an AAV virus carrying the HEXA gene; the first successful results were reported in 2022.
Other lysosomal storage diseases are due to deficiencies in genes encoding proteins needed for protein targeting to lysosomes, or for maintaining the lysosomal ionic environment. One example is I-cell disease, a deficiency of the Golgi phosphotransferase needed to produce terminal mannose-6-phosphate.
Protein turnover and amino acid reutilization
Some proteins are normally rapidly tagged with ubiquitin for degradation and have short half-lives unless this process is inhibited; others are ubiquitylated in response to activation of an E3 ligase. Most proteins are marked for degradation when they lose their native structure due to denaturation, proteolysis, or accumulation of structural modifications. The 20 amino acids are subject to more than 200 different post-translational modifications, some of which signal that a protein has aged and needs to be replaced. These include oxidation of methionine, deamidation of asparagine, and glycation (non-enzymatic attachment of sugar residues) of primary amines (on lysine side chains and terminal amino groups of proteins). This surveillance system helps rid the cell of dysfunctional proteins that might also form toxic aggregates.
Protein degradation is also a source of amino acids for the production of new proteins or as a source of energy. In the latter case, the amino groups must be removed to make urea for excretion, and the carbon skeleton feeds into the TCA cycle in various ways, generating reduced electron carriers for the ETC to generate ATP, or is used to make citrate that is exported from the mitochondria for fatty acid biosynthesis and energy storage. In a normal 70 kg adult, ~300g protein is synthesized each day. This requires an oral protein intake of just 70g/day, because most of the new protein is made from recycled amino acids. Think about that the next time you eat a 16 oz steak (120 g protein) or drink a “protein shake” (100 g protein/8oz glass!) Any additional amino acids beyond what is needed for protein synthesis must be converted to energy or fat, and in both cases the liver and kidneys must get rid of the nitrogen.
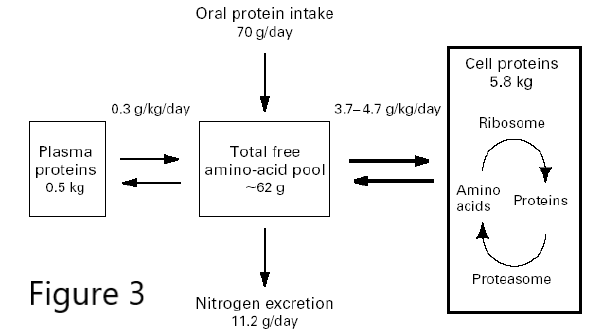
Figure 3. Bulk Protein turnover.
Excess protein degradation, and therefore nitrogen excretion, can become a problem, not just when too much is eaten, but also during prolonged fasting or starvation, when muscle protein must be degraded to provide glucogenic amino acids for gluconeogenesis. It can also occur in certain diseases, like AIDS and cancer, when large numbers of cells are undergoing apoptosis and their proteins are degraded. Clinically, this muscle wasting is called cachexia, and it is seen in the late stages of almost every major chronic illness, affecting many people with heart failure, chronic obstructive pulmonary disease and kidney disease. It is driven in part by inflammatory signals that activate expression of ubiquitin E3 ligases, leading to protein degradation.
Feedback: