
7 Cholesterol, Lipid Transport
Introduction: Lipids are commonly referred to as fats. Lipids are large hydrophophobic molecules that do not dissolve in water. Fatty acids (long chain carbon molecules that end in a carboxyl group) are key components of the lipid bilayer of the cell membrane and intracellular organelles. Lipids play a key role in regulating transport of molecules across extracellular and intracellular compartments. Fatty acids and triglycerides (three fatty acids attached to a glycerol backbone) are a major source of energy for the body, including the heart and skeletal muscle. Specialized functions of lipids include the development of surfactant in the lung, formation of bile to facilitate the transport and absorption of dietary lipids in the gut, insulating nerves as myelin, to name only a few. Lipids also serve as signaling molecules, serving as targets of lipid kinases and substrates for eicosanoid signals. The lipid cholesterol (a large molecule with multiple 6 and 5 carbon rings) is a structural component of all cell membranes and is a precursor to bile acids, and steroid (molecules synthesized from cholesterol) hormones including vitamin D. Appropriate cellular levels of cholesterol are essential for normal function. To ensure this, the body obtains cholesterol from the diet and synthesizes cholesterol from other molecules. Dysregulation of cholesterol transport pathways leads to hypercholesterolemia and contributes to atherosclerosis.
This session focuses mostly on lipid physiology outside the cell. We introduce key concepts related to the digestion, absorption and transport of lipids (fats). What are micelles? What is the role of bile salts? What are possible causes of lipid malabsorption? What are the key differences between the exogenous and endogenous pathway of lipoprotein transport? What is a key anatomical difference between lipid and carbohydrate absorption from the gut? How are circulating triglycerides absorbed? What is unique about peripheral cholesterol metabolism?
This information will connect with subsequent blocks that introduce you to gastroenterology and endocrinology
Session Learning Objectives and Brief Synopses (see main text for detailed explanations):
Session Learning Objective 1. Outline the pathways of lipid digestion and transport in the body, including the roles of chylomicrons, VLDL, IDL, LDL, and HDL.
Session Learning Objective 2. Outline pathways of dietary cholesterol uptake and transport.
Lipid Digestion and Transport Overview: Dietary (exogenous) lipids (primarily triglycerides) are broken down in the gut because these large molecules cannot cross the cell membrane. Digestion of triglycerides requires bile acids (made by the liver), which act as detergents, and pancreatic enzymes (lipases). The resulting fatty acids are taken up by the mucosal cells of the small intestine, converted back to triglycerides, and secreted into the lymphatic system. Because of poor solubility in lymph or plasma (mostly water), lipids are assembled into soluble lipoprotein particles (see immediately below) for transport and for specific delivery to target tissues. Triglycerides are also synthesized in the liver (endogenous lipids, assembled from serum fatty acids or made from Acetyl CoA) and are secreted into the hepatic vein. Finally, a system exists for transport of extrahepatic lipids (mostly cholesterol) from peripheral tissues to the liver (see legend for a summary).
Lipoprotein Particle Overview: Most lipids are not soluble/miscible in water (aqueous, plasma). To be transported in blood, lipids are assembled into lipoprotein particles (see Table 1). These particles are mini lipid droplets with a surface of phospholipids (the hydrophilic, charged (polar), phospho head groups of the molecules on the outside, the hydrophobic fatty acid tails on the inside – much like the outer leaflet of a cell plasma membrane). Buried inside (the core) are all the hydrophobic triglycerides and cholesterol esters (and a little free cholesterol). Key large proteins (apolipoproteins) serve as scaffolds for assembly and maintaining particle integrity, but also are recognized by cell surface receptors (eg. ApoE receptor and Apo B-100 receptor) and serve to activate key enzymes (Apo C activates LPL, Apo A activates LCAT, more below). The nomenclature of lipoproteins is archaic based on their separation when centrifuged in a density gradient, while the lipid content and surface proteins are a much more important definition of their role than the density.
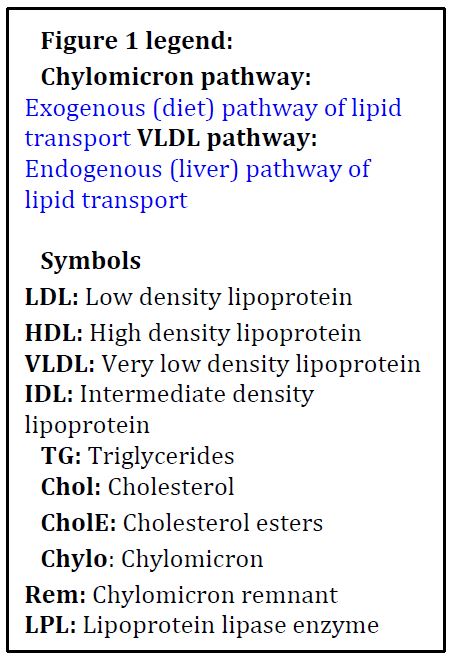

Figure 1: Schematic of Exogenous and Endogenous Lipoprotein Cycles
Clinical Overview: Most clinicians only relate to lipid metabolism in the context of risk of coronary artery disease. LDL (‘bad’ cholesterol) is the longest lasting lipoprotein particle and if present in excess or modified (eg. oxidation) is deposited within macrophages in the walls of arteries leading to atherosclerotic plaques. Similarly, low levels of HDL (‘good’ cholesterol) results in atherosclerosis via reduced transport of cholesterol from peripheral tissues back to the liver. Altered lipoprotein metabolism is usually caused by a complex interaction of nutrition, activity levels, polygenic inheritance and environmental factors. However, rare monogenic disorders increase the risk of cardiovascular disease by altering lipid metabolism at the level of lipoprotein secretion and uptake from plasma. An understanding of lipolysis in white adipose tissue is also important to understanding ketoacidosis, a life-threatening condition most commonly associated with diabetes mellitus.
Board Exam Overview: In addition to the above, there are extremely rare monogenic disorders that impair lipid metabolism at the level of intracellular transport and mitochondrial fatty acid degradation (beta oxidation). These disorders almost always present in childhood as low energy states (weakness, fatigue, failure to thrive, hypoglycemia) due to inadequate cellular fuel/ATP production. It is important for you to be able to distinguish these from glycogen storage disorders with a similar presentation (hint: always look for the ketones! If ketones are present it is NOT a problem with fatty acid transport/oxidation).

Table 1: Important characteristics of lipoprotein particles. These particle vary dramatically in 1) size; 2) the apolipoproteins present on the surface of the particle – important for uptake by specific receptors and activating specific enzymes; 3) the triglyceride (TG) versus the cholesterol ester and cholesterol (CE + C) content inside the particle core, and protein content on the surface.
Dietary Lipids
On average, North American adults consume 150 g of triglyceride, 300mg of cholesterol and 4-8 g of phospholipid daily. Fats aggregate as lipid droplets in water, and your body needs to make these droplets as small as possible to optimize digestion and absorption. Multiple gut specific physical mechanisms contribute (peristalsis, pyloric opening, etc.), but the most important step is chemical. In the small intestine, fats interact with bile salts (produced by the liver, stored in the gallbladder, empty into the gut through the pancreatic duct following fatty meals in response to various gut hormones) which act as powerful detergents and separate the fat droplets into micelles (tiny droplets). Micelles hugely increase the surface area of fat exposed to pancreatic lipases, which digest the triglycerides into fatty acids and monoacylglyerol.
Intestinal mucosal cells absorb fatty acids, cholesterol, monoacylglycerols, and phospholipids from the gut lumen into the cell interior by poorly understood mechanisms (FA and C can diffuse into the cell, but the charged molecules cannot). Within the mucosal cells, triglycerides are synthesized from monoacylglycerols and fatty acids, and cholesterol is re-esterified with a long chain fatty acid by acyl CoA cholesterol acyl transferase (ACAT) (Figure 2). Some short-chain and medium-chain fatty acids (usually fewer than 10 carbons) are secreted directly into the portal system and are not assembled back into triglycerides.
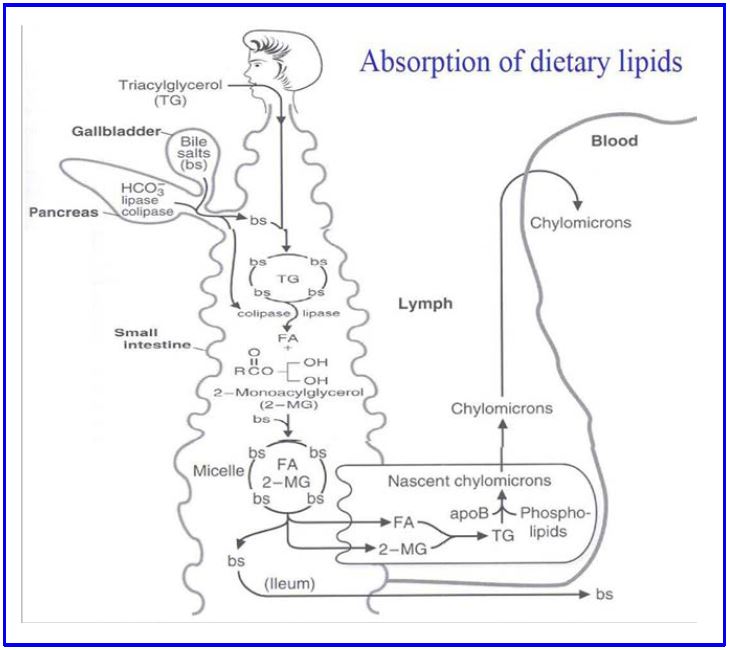
Figure 2: Absorption of dietary lipids
Figure 2 Legend: TG: triglycerides, BS: bile salts, FA: fatty acids, 2-MG: 2-monoacyl glycerol, ApoB: Apo-B48 apolipoprotein, HCO3–: bicarbonate.
Clinical questions. What happens if your pancreas is damaged and can’t make enough lipase? Can pancreatic enzymes be replaced by medications? What happens if bile salts and ingested lipids don’t mix in the right quantity or the right location? Does gallbladder surgery (cholecystectomy) cause this? Do gallstones (choledocholithiasis) cause this? What is the effect of medications that bind and sequester bile salts (bile acid sequestering resins)? What happens to stool (feces) if fat is not digested and absorbed? Can you prescribe medications that block intestinal fat absorption?
Test your understanding of lipid digestion and absorption with the following exercise. Rouy-en-Y gastric bypass is a common surgical weight loss procedure that bypasses most of the stomach by attaching the jejunum to a limited gastric pouch (see diagram, you will not be tested on any details of this procedure in this block)
Exogenous Lipid Cycle
Assembly and Secretion
The lipoprotein particle secreted from intestinal cells is called a chylomicron which is how the gut delivers lipids to the rest of the body (see Table 1). The chylomicron is assembled in the rough endoplasmic reticulum starting with the ApoB48 apolipoprotein being imported into the ER. ApoB is a 29 exon gene that makes a giant 4563 amino acid protein. In intestinal cells specific RNA editing creates a premature stop codon so only 48% of the normal ApoB protein is translated (hence ApoB48). Further processing of the chylomicron occurs in the golgi. The entire process takes time (1-4 hours after eating) and costs energy (diet-induced thermogenesis). Chylomicrons are very large (>100nM) and contain mostly triglycerides (86%)(Table 1, Fig 3).
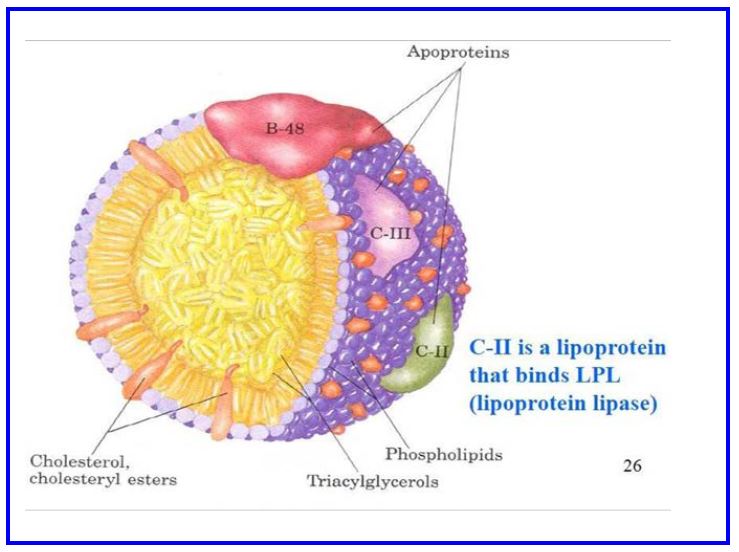
Figure 3: Schematic diagram of a chylomicron
Chylomicrons are secreted from intestinal cells into retroperitoneal lymphatic lacteals that eventually drain into the subclavian vein through the thoracic duct (Fig. 2). This route intentionally avoids the liver initially (unlike carbohydrates and amino acids which enter the portal vein) so other tissues have the first chance to remove triglycerides (and cholesterol and phospholipids) from the chylomicron (Fig. 4). The secreted chylomicron also has Apo A on the surface (mostly transferred rapidly to HDL particles). Once out of lymph and in the blood, chylomicrons rapidly acquire Apo C and Apo E by bumping into already circulating lipoproteins. ApoC activates an enzyme called lipoprotein lipase (LPL) on the surface of endothelial cells and allows for triglycerides inside the chylomicron to be hydrolyzed into fatty acids and monoacylglycerol for cellular absorption. Apo E has high affinity for a hepatic receptor which takes up chylomicron remnants. Because of this chylomicrons are cleared from plasma rapidly and the estimated residence time in plasma of any individual chylomicron particle is only 5-10 minutes.
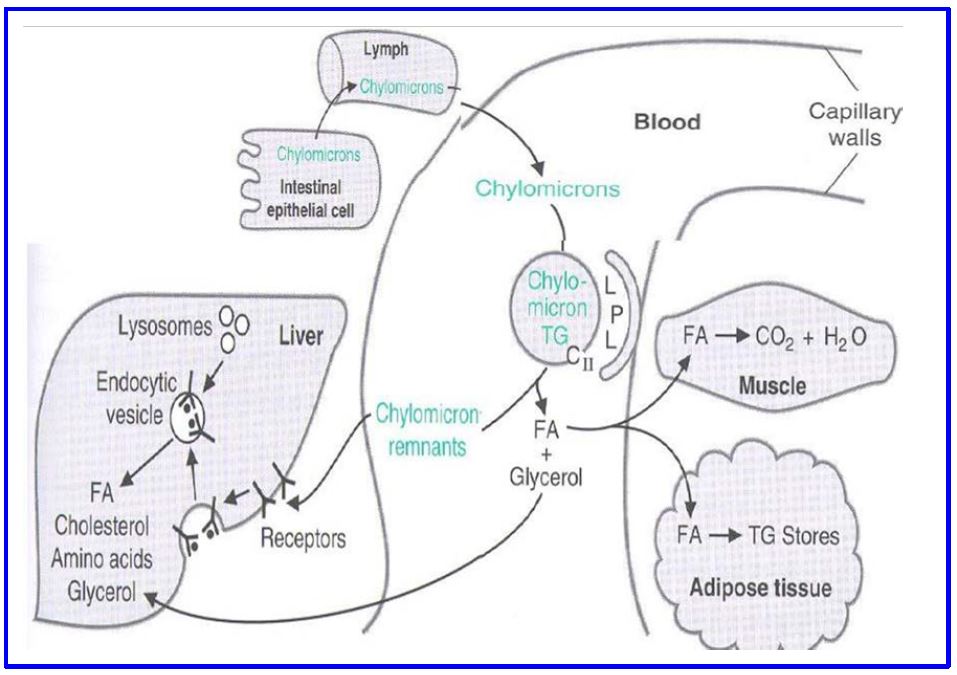
Figure 4: Fate of chylomicrons
Legend. Fig 4 shows the production and fate of chylomicrons and the lipids contained in these gut-derived particles
Delivery of triglycerides. Lipoprotein lipase (LPL) is an enzyme present on the exterior surface of capillary endothelial cells loosely bound to a glycoprotein molecule called heparan sulfate. The enzyme is made by cells within these tissues (eg. myocyte, adipocyte) and secreted into the local capillaries. LPL is most abundant in adipose tissue (including mammary gland), where its synthesis is increased by insulin signaling, but also present in skeletal muscle and heart muscle and at lower levels in many tissues. Triglycerides within circulating chylomicrons is hydrolyzed by LPL and the products (a monoacylglycerol and two fatty acids) (Fig. 4) are taken up by those local cells (eg. adipocytes) and resynthesized into triglyceride, or used as a substrate for energy metabolism by other tissues such as cardiac and skeletal muscle, or converted to phospholipids to enter a lipid bilayer. One of the apolipoproteins of the chylomicron (APO-CII) is an activator of LPL. The remains of the chylomicron after LPL action to remove triglycerides are called chylomicron remnants.
Cellular Uptake. At least two receptors on the hepatocyte cell surface bind and internalize chylomicron remnants (Fig. 6; below). Binding to both receptors is mediated by interaction with APO-E. The LDL receptor (LDLR, Apo-B100/E receptor) is a major pathway because of its strong affinity for Apo-E containing lipoproteins. However, this is not the only receptor because chylomicron remnants do not accumulate in the plasma of LDL receptor-defective individuals. The second mechanism involves LDL receptor-related protein (LRP). Chylomicron remnants are internalized by receptor-mediated endocytosis. The lipid of the remnant (glycerophospholipid, cholesterol and some triglyceride) is made available for metabolism by the hepatocyte. Chylomicrons also carry dietary lipids such as β-carotene and the fat-soluble vitamins (vitamins A, D, E, and K), which are stored in the liver for systemic use.
Session Learning Objective 3. Understand the possible mechanisms of familial hypertriglyceridemia.
Clinical. Hypertriglyceridemia is a common diagnosis. Mildly elevated TG levels (150-500 mg/dL) can contribute to cardiovascular disease risk. Severely elevated TG (>1000 mg/dL) can cause acute pancreatitis and causes characteristic lipid deposits in skin (xanthelasma (smaller) and xanthomas (larger)). Based on the above you should be able to predict rare monogenic mutations that result in high serum triglycerides (hypertriglyceridemia). Inactivating mutations in the LPL gene present in childhood. Frequency is 1 per million. Mutations in Apo C-II can reduce the ability to activate LPL. Apo C-II deficiency is extremely rare. Three common Apo E alleles are present in all populations (E2, E3, E4). Apo E2 has a single aa substitution (Arg158-Cys) from Apo E3 and has much lower affinity for the LDL receptor. Homozygosity for Apo E2/E2 is present in about 0.5% of studied populations. Homozygosity results in mild hypertriglyceridemia but can become severe when other conditions are superimposed, including fatty liver, diabetes mellitus, and various medications (estrogen, retinoic acid medications). Fibrates are the most common medication used to reduce triglyceride levels. These medications activate a key family of transcription factors (peroxisome proliferator activated receptors (PPARs)) that reduce triglycerides at multiple levels of lipid metabolism.
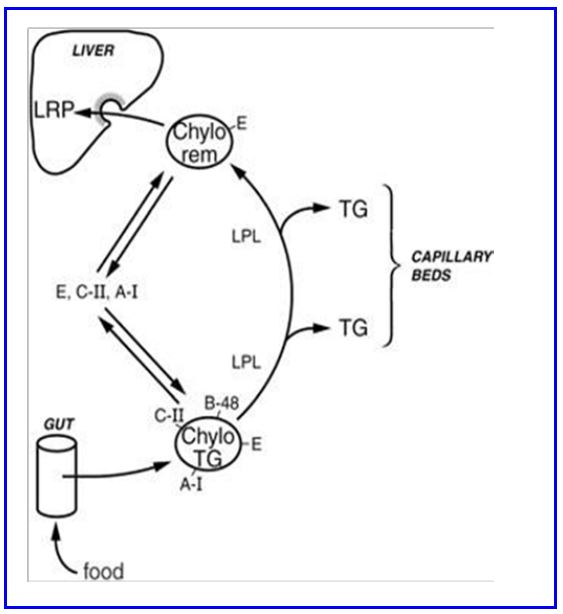
Figure 5
Simplification. As usual the full truth is much more complicated. Lipid absorption and tissue/organelle fatty acid targeting is incredibly complex. Absorbed fatty acids are rapidly and efficiently partitioned and rare fatty acids (will be discussed later) are preferentially turned into phospholipids rather than assembled into triglycerides or metabolized. Specialized plant derived and animal derived fatty acids have unique signaling mechanisms. This point is highlighted by the ability of fish oils (specific omega-3 fatty acids) to affect lipid metabolism and lower serum triglyceride levels. The mechanism is complex, but the best documented is an effect of fish oils to decrease the delivery of free fatty acids to the liver by decreasing triglyceride hydrolysis in adipocytes. This has an effect to decrease VLDL production by the liver.
Endogenous Lipid Cycle
Your 8 year old nephew only eats plain pasta. No butter. His circulating lipids are normal. Why? His liver happily takes all the glucose from pasta (carbohydrate) and turns it into triglycerides and cholesterol (fats) and exports those lipids in periods between meals. The production of very low density lipoprotein (VLDL) by the liver is how hepatic triglycerides and cholesterol is delivered to peripheral tissues. The rates of cholesterol delivery to plasma (VLDL is to the liver what chylomicrons are to the gut) and receptor mediated uptake of VLDL, IDL, and LDL determine the level of plasma LDL cholesterol (Fig 6; below).
Assembly and secretion. The liver has cytoplasmic enzymes that can make fatty acids or cholesterol starting from acetyl CoA (details covered in lipid biosynthesis session). These endogenous fatty acids can be elongated and desaturated if necessary within the ER. Fatty acids are linked to glycerol (3 C sugar) to form triglycerides. VLDL particles are assembled in the ER and golgi (similar to chylomicrons in the intestinal cells) except the major scaffold lipoprotein is the full length ApoB100. The process of hepatic VLDL production is accelerated dramatically by hepatic uptake of fatty acids from adipocytes (released from triglycerides through activation of hormone sensitive lipase in fasting). VLDL is secreted into the hepatic venous system (not into lymphatics). Once in the blood VLDL acquires Apo C and Apo E. VLDL does not have Apo A to donate to HDL particles. VLDL is secreted from the liver in low amounts continuously, but the rate increases in the post-prandial state (when insulin signaling returns to fasting levels and adipocyte lipolysis increases). This is likely because cells prefer to get their supply of fatty acids for fuel from circulating lipoprotein particles rather than having higher levels of (free, meaning carried by albumin) fatty acids circulating in the blood.
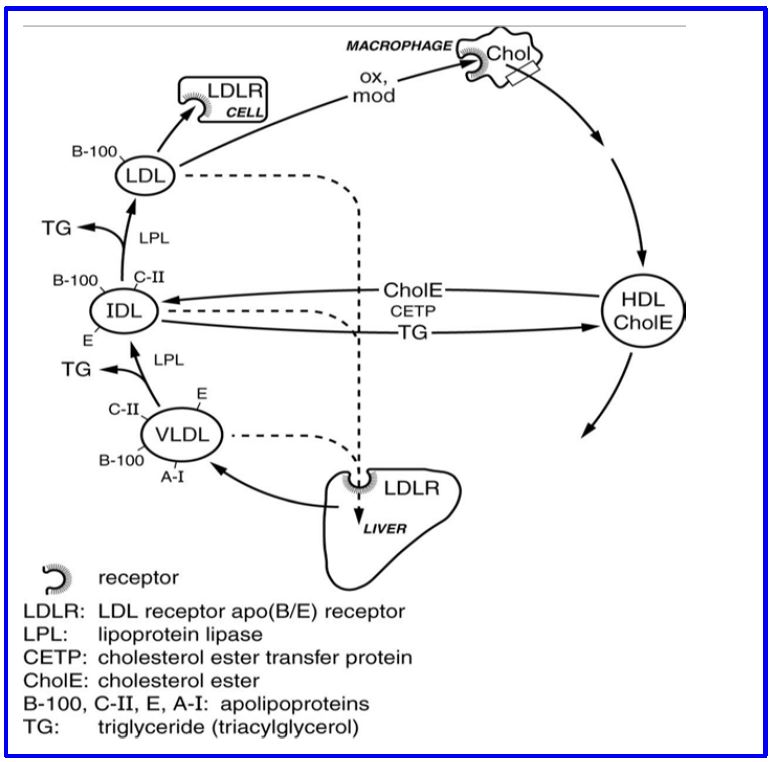
Figure 6: The VLDL transport pathway of endogenous lipids
Delivery of triglycerides. Similar to chylomicrons, lipoprotein lipase (LPL) activity results in the hydrolysis and removal of VLDL triglyceride in the vascular bed of different tissues. Hepatic lipase, a triglyceride and glycerophospholipid lipase, also plays a role in removal of triglyceride from VLDL and IDL. The resulting smaller lipoprotein, called intermediate density lipoprotein (IDL), can continue to circulate and be metabolized further to an LDL. IDL is also removed from circulation by receptors on the surface of hepatocytes and other cells that recognize Apo E. When IDL loses all the surface ApoE (and most internal triglyceride) it becomes low density lipoprotein (LDL). This particle circulates the longest in plasma, because it only has Apo B100 which has much lower affinity than Apo E for the LDL receptor and so is taken up very poorly by the liver. This long half-life contributes to making LDL the ‘bad’ cholesterol because it is more likely to become damaged (eg. oxidized) and more likely to be mistakenly taken up by receptors on peripheral cells (scavenger receptors, eg. SRB1, on many cells, but most importantly on monocyte/macrophages).
Cellular uptake. Like chylomicron remnants, receptor-mediated endocytosis is how IDL and LDL are taken up by cells. Uptake by hepatocytes accounts for about half of LDL endocytosis in humans, but most cells express the LDL receptor (at lower levels than hepatocytes) and can take up LDL particles. In plasma, the loss of Apo E from the surface of the particle (IDL becomes LDL) is a critical factor in the rate of uptake of lipoprotein particles by the LDL receptor (lower affinity for Apo B-100 than Apo E). This contributes to a particle half-life for LDL of about 3 days. Once endocytosed the LDL/LDLR complex traffics to the lysosomal compartment. The LDL receptor can be either degraded or recycled to the cell surface (see Clinical content below). The endocytosed lipoprotein contents (phospholipids, cholesterol and (scant) triglycerides) are metabolized or stored by the cell. Complex intracellular signals recognize intracellular cholesterol stores and control LDL receptor gene expression and the quantity of LDL receptors on the cell surface (important for pharmacological therapy).
Exchange of lipids with HDL. In the process of conversion of VLDL to IDL and then to LDL (the archaic nomenclature is confusing here, IDL is intermediate between VLDL and LDL. Not between LDL and HDL) there is also exchange of cholesterol ester for triglyceride which occurs when these particles bump into HDL lipoproteins (see below). This exchange is mediated by cholesterol ester transfer protein (CETP, a liver protein that circulates mostly bound to HDL) (Fig 6; above). The triglyceride composition of the IDL and LDL decreases and the cholesterol ester content increases as a result of this exchange.
Session Learning Objective 4. Understand the biological basis of familial hypercholesterolemia.
Clinical. The clinical scenario highlighted in this section is familial hypercholesterolemia (FH). This disorder is caused by inactivating mutations in the LDL receptor that decrease the uptake of LDL from plasma (variety of mutations described that affect affinity for Apo B100, receptor production or receptor recycling). Homozygous patients are rare, and present with cardiovascular disease at a very early age (< age 30) and very high plasma LDL levels. Heterozygotes have an intermediate plasma LDL phenotype with early onset cardiovascular disease (age <50). Heterozygote frequency can be as high as 1/250 in some populations. At the end of this chapter, you should be able to describe how treatment with statins (HMG CoA reductase inhibitors) can decrease LDL levels in patients with FH.
Clinical content. A powerful novel treatment for patients with FH are PCSK9 inhibitors. PCSK9 is a hepatic proprotein convertase present in plasma. It binds to the extracellular portion of the LDLR on the cell surface. When bound, PCSK9 targets LDLR for degradation in the lysosome after the LDL/LDLR complex is endocytosed. PCSK9 inhibitors prevent PCSK9 from binding to LDLR and dramatically increase LDLR recycling back to the cell surface – causing an extremely powerful effect to reduce plasma LDL.
Reverse Cholesterol Transport. This is the most complex of the three lipoprotein cycles covered in this course pack. The main lipoprotein for reverse cholesterol transport is high density lipoprotein (HDL). The key apolipoprotein on the surface of HDL is Apo A. Liver (70%), small intestine (20%) and other tissues (10%) can all make HDL. Cholesterol (an essential part of cell membranes and organelles) is very poorly metabolized by most cells. When cells accumulate too much cholesterol or die (apoptosis, necrosis, etc.), free cholesterol from those cells needs to be removed. HDL does that job.
Assembly and secretion. HDL is secreted as a small flat disk with Apo A and phospholipids (like the very initial stage of chylomicrons or VLDL in the rough ER), but otherwise empty (nascent HDL). The half-life of HDL is up to 24 hours (long by lipoprotein standards). HDL bumps into other lipoproteins and acquires Apo E (but is not taken up by the LDL receptor, mostly exchanges this back to chylomicrons and VLDL) and Apo C (may deliver some TG to tissues by activating LPL, but mostly gives Apo E back to chylomicrons and VLDL), and more Apo A (from chylomicrons).
Delivery of cholesterol to HDL. Very complex. Multiple mechanisms exist. The best described mechanism involves ATP binding cassette proteins (eg. ABCA1, ABCG1) which function as cholesterol pumps from the cell surface into HDL particles. On the HDL particle surface, the enzyme lecithin cholesterol acyl transferase (LCAT, synthesized in liver, bound to HDL) immediately converts free cholesterol to an acylated cholesterol ester. LCAT activity is increased by more Apo A. The cholesterol ester begins to fill up the empty HDL interior. Other mechanisms may involve HDL endocytosis, intracellular cholesterol loading and then reverse endocytosis.
Cellular uptake of HDL. Again, complex. While still circulating, HDL transfers cholesterol esters to IDL and LDL (via CETP activity on the HDL surface, see above). This is one mechanism whereby peripheral cholesterol enters the liver to be recycled (as bile salts or secreted into bile) or re-secreted in VLDL. HDL is also recognized by the scavenger receptor B1 (SRB1) on hepatocytes which (by endocytosis or cholesterol transfer) is another way HDL delivers peripheral cholesterol to the liver. HDL is also taken up by other cells, for example glands that make cholesterol based steroid hormones (adrenal, ovary, testes) can take up HDL when these glands need cholesterol (they also make their own cholesterol, see below).
Cholesterol Excretion. Most cholesterol recycled from peripheral tissues is excreted into and eliminated by the gut. About 65% comes directly from hepatocytes secreting cholesterol taken up from HDL (directly or indirectly) as free cholesterol in bile. Less well understood, about 35% of cholesterol excretion happens directly in the gut (TICE, trans-intestinal cholesterol efflux). These processes involve two key nuclear hormone receptors LXR (liver X receptor) and FXR (farnesoid X receptor) that transcriptionally control a variety of cholesterol transporters and bile synthesis enzymes. The cholesterol modified into bile salts by the liver and secreted into the bile does not count because bile salts are 90+% reabsorbed in the distal small intestine.
Clinical. Two clinical vignettes relate to this section.
First, Tangier disease is a very rare autosomal recessive disease causing loss of function mutations in ABCA1. This results in extremely low serum HDL levels and impaired reverse cholesterol transport. Cholesterol esters accumulate in the reticuloendothelial system of many cells. Tangier disease causes early cardiovascular disease.
Second, clinical and pre-clinical studies showed that mutations that reduced CETP function were associated with higher HDL levels, increased reverse cholesterol transport, lower LDL levels and reduced cardiovascular events. Pharmaceutical companies developed CETP inhibitors, but multiple clinical trials demonstrated no reduction in cardiovascular disease despite significantly increased HDL levels with these medications.
Session Learning Objective 5. Outline the major pathway of de novo cholesterol synthesis. Understand the biochemical basis of statin action.
Cholesterol Synthesis
This section takes you from lipid particles within plasma back to lipid synthesis inside the cell. We already learned about cytoplasmic regulation of fatty acid synthesis (from Acetyl CoA indirectly leaving the mitochondria via citrate). Most cells can also make their own cholesterol which is critical because cholesterol is needed for normal plasma membrane function (among other uses). Like fatty acids, production of cholesterol occurs in the cytoplasm.
Biosynthesis of Cholesterol:
The structure and carbon numbering of cholesterol are shown in Fig. 7. The first steps in cholesterol synthesis are called the mevalonate pathway.
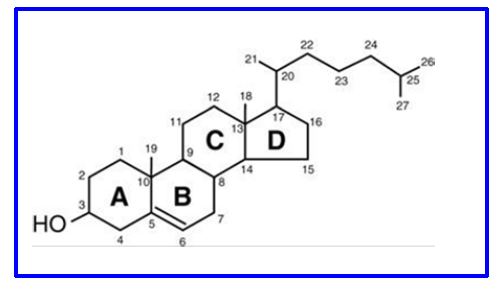
Figure 7. Structure of cholesterol
Mevalonate pathway. This pathway starts with two acetyl-CoA molecules condensing to form acetoacetyl-CoA. In the second step another acetyl CoA is added to form HMG-CoA (C6). The third step is the rate limiting step when HMG-CoA is reduced to form mevalonate. This reduction requires two NADPH and is mediated by the enzyme HMG-CoA reductase. This step is tightly controlled by transcriptional control of the enzyme and by covalent regulation (see below). Finally, mevalonate is decarboxylated and has a pyrophosphate group added in multiple steps requiring 3 ATP to form the C5 molecule isopentenyl pyrophosphate (isoprenoid subunit).

Figure 8. Overview of Cholesterol synthesis
You have already seen some of these enzymatic steps leading to HMG-CoA in hepatocyte mitochondria related to ketone production. The enzymes responsible for HMG-CoA production starting cholesterol synthesis in the cytoplasm are a different set of enzymes.
Regulation of HMG-CoA reductase activity
Covalent Control of cholesterol biosynthesis. HMG-CoA reductase is inactivated via phosphorylation by cAMP dependent protein kinases; dephosphorylation by protein phosphatases reactivates (Figure 9), making cholesterol biosynthesis subject to hormonal control. In addition, cholesterol binds a nuclear receptor that inhibits transcription of the reductase gene. This is an example of the end product of a biosynthetic pathway inhibiting the key committed step.
Clinical. Statins: HMG-CoA reductase inhibitors are very commonly prescribed medications to lower serum LDL levels and reduce the risk of cardiovascular disease. Common examples include atorvastatin and simvastatin. The medications reduce circulating LDL in at least two ways. First, inhibition of HMG-CoA reductase directly reduces the output of the cholesterol biosynthetic pathway in all cells. In the liver this reduces the release of VLDL into the circulation reducing eventual LDL particles. Second, in addition, decreased cellular cholesterol concentration in all cells upregulates the transcription of the LDL receptor gene, and LDL receptor expression on the cell surface. This increases clearance of LDL from plasma. Side effects of statins include muscle aches, and hepatitis, at least partly related to inadequate intracellular cholesterol levels.
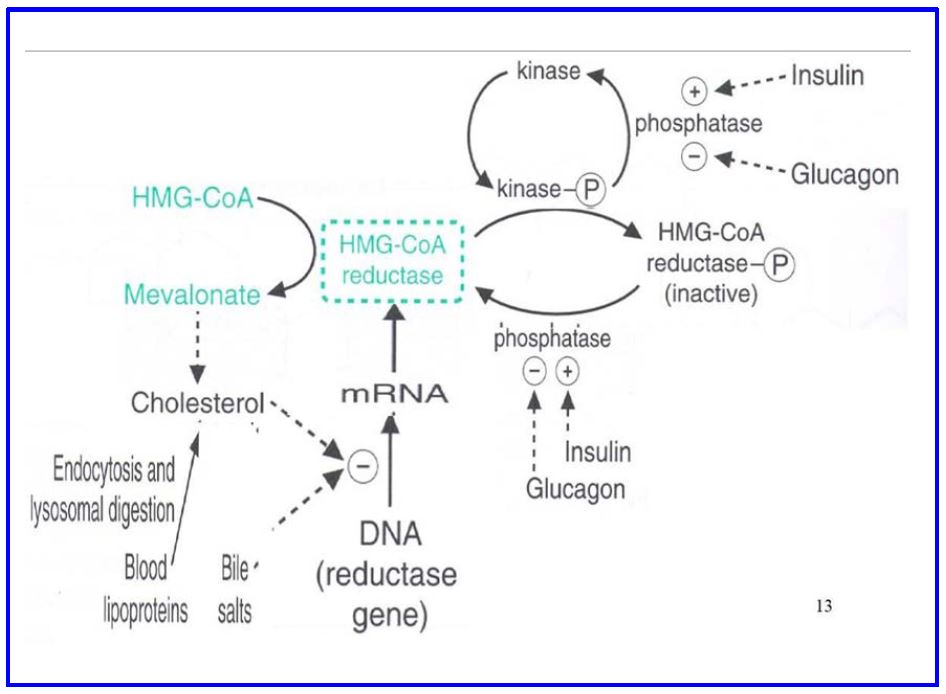
Figure 9 Regulation of Cholesterol Synthesis
Subsequent steps in Cholesterol Synthesis: Multiple complex enzymatic steps result in the polymerization and cyclization of 6 isoprenoid units (C30 molecule called squalene) and then demethylation and migration of double bonds eventually yields cholesterol (C27).
Cholesterol can be esterified at the number 3 carbon with long chain fatty acids (e.g., oleate, linoleate). This molecule is more hydrophilic, easier to transport in plasma, and is not a membrane structural lipid. Note that the planar structure of Fig. 7 does not represent the 3D shape of molecular cholesterol.
Isoprenoid subunits: These C5 building blocks are important for the structure of many molecules including vitamin A and E, the mammalian hormone Vitamin D, as well as carotenes and xanthophylls, which have effects on immune function among other effects.
Review questions:
1. What are chylomicrons used for?
2. How does the pathway for endogenous lipid transport differ from the exogenous pathway?
3. What is the role of lipoprotein lipase? Where is it found?
4. Explain the function of bile salts.
5. Explain the function of the LCAT enzyme
6. What are the functions of the Apo C, Apo E, Apo A and Apo-B100 lipoproteins?
7. What is the rate limiting step of cholesterol biosynthesis?
8. Explain the diverse mechanisms by which the activity of HMG-CoA reductase is regulated.
9. How do statins affect cholesterol biosynthesis?
10. Outline the key steps of the de novo cholesterol biosynthesis pathway
Feedback