
8 Lipid Metabolism, Lipid Disorders, Arachidonic Acid Metabolism
This Pressbook covers three topics, each with its own session learning objectives: and 1) lipid metabolism, 2) ketone bodies and 3) Arachidonic Acid and Eicosanoids.
Fatty acids are important for the formation of cell membrane bilayers and intracellular organelles and are key elements for regulating transport of molecules across extracellular and intracellular compartments. Fatty acids and triglycerides are a major source of energy for the body, including the heart and skeletal muscle. Specialized functions of lipids include the development of surfactant in the lung, formation of bile to facilitate the transport and absorption of dietary lipids in the gut, insulating nerves as myelin, to name only a few. Lipids also serve as signaling molecules, serving as targets of lipid kinases and substrates for eicosanoid signals. The lipid cholesterol is a structural component of all cell membranes and is a precursor to bile acids and steroid hormones including vitamin D. Appropriate cellular levels of cholesterol are essential for normal function. To ensure this, the body obtains cholesterol from the diet and synthesizes cholesterol in a carefully regulated series of steps. Fatty acids and cholesterol are shuttled around the body in large lipoprotein particles. Disorders of lipid metabolism are involved in major clinical disorders including atherosclerosis, diabetes, and obesity.
Session Learning Objectives and Brief Overview
— fatty acids enter the mitochondrial matrix through the carnitine shuttle and are oxidized to acetyl CoA by specific acyl CoA dehydrogenases, NADH is produced directly by the oxidation cycles and through the TCA cycle. ATP production is high compared to carbohydrate metabolism.
— white adipose tissue stores triglycerides in large intracellular lipid droplets. Insulin stimulates TG storage. Low insulin levels and counterregulatory hormones stimulate hormone sensitive lipase (HSL) which degrades TG into free fatty acids and glycerol.
Session Learning Objective 3. Outline the pathway and regulation of de novo lipid biosynthesis.
— high cellular energy allows for production of fatty acids in the cytoplasm. Starting from mitochondrial intermediates that need to be shuttled into the cytoplasm.
— ketones are produced from acetyl CoA and released by the liver during fasting. Many tissues take up and metabolize ketones for energy.
— Clinical SLO. Monogenic disorders exist that impair beta oxidation and present as failure to thrive in infants/children.
— understand the initial steps in the arachidonic acid pathway including the role of phospholipase A2, COX-1 and COX-2 enzymes and the medications that inhibit these steps.
— understand clinical examples of the roles of specific prostaglandins and thromboxanes in causing symptomatic disease.
Fatty Acid Oxidation Overview. Fatty acid oxidation is a key source of ATP to fuel cellular energy needs. Fatty acids are stored as components of triglycerides (TG) in adipose tissue and released into the blood in response to epinephrine. Most cells (not neurons, not red blood cells) can use fatty acids for fuel. In exercising muscle, fatty acids are oxidized to acetyl CoA, which enters the TCA cycle to generate ATP. In the liver, ATP derived from fatty acid oxidation is used as an energy source for gluconeogenesis. The liver converts fatty acid derived acetyl CoA to ketones, which can be used by many cells (including neurons after a period of adaptation) as a source of ATP production.
Fatty acid synthesis Overview: Fatty acids (mostly C16 palmitate) are synthesized from acetyl CoA in the cytoplasm. The main tissues where fatty acid synthesis occurs are liver and adipose tissue, although synthesis can occur in almost all tissues. Control of the biosynthetic pathway occurs at the first committed step: the synthesis of malonyl CoA, catalyzed by acetyl CoA carboxylase. In addition to its role as a substrate for fatty acid biosynthesis, malonyl CoA plays a key role in coordinating fatty oxidation and synthesis. The synthesized palmitate can be modified within the cell. Enzymes of the endoplasmic reticulum can introduce double bonds and elongate beyond 16 carbons (palmitate). Synthesized fatty acids can be esterified to glycerol and stored as triglycerides. Many tissues also use synthesized fatty acids to make phospholipids. Insulin is the main hormone that promotes synthesis of fatty acids and triglycerides.
Eicosanoid Overview: Arachidonate (a C20:4, Δ5,8,11,14 polyunsaturated fatty acid) is a precursor for a group of complex lipids that mediate numerous biological processes. Collectively, these substances are termed eicosanoids and serve as a communication pathway between cells that can alert surrounding cells related to cell damage and inflammation. The biosynthetic pathway begins with the release of arachidonate from glycerophospholipids in the inner leaflet of the plasma membrane, catalyzed by the enzyme phospholipase A2. Further metabolism of arachidonate is accomplished by at least different chemical reactions in various tissues resulting in production of prostaglandins, leukotrienes, and lipoxins. Because of their key roles as inflammatory mediators, medications have been developed that alter eicosanoid signaling.
Session Learning Objective 1. Analyze the oxidation of fatty acids, including pathway regulation, the carnitine shuttle and generation of ATP.
Fatty Acid Oxidation
Goals: 1. Appreciate different classes of fatty acids; 2. Analyze the oxidation of fatty acids and explain why it yields more energy than catabolism of carbohydrates or proteins. 3. Explain how fatty acids are transported into the inner mitochondrial matrix and activated for beta-oxidation. 4. Explain how fatty acids are mobilized from adipose stores; 5. Be cognizant of the difference between beta-oxidation of odd carbon versus even carbon fatty acids.
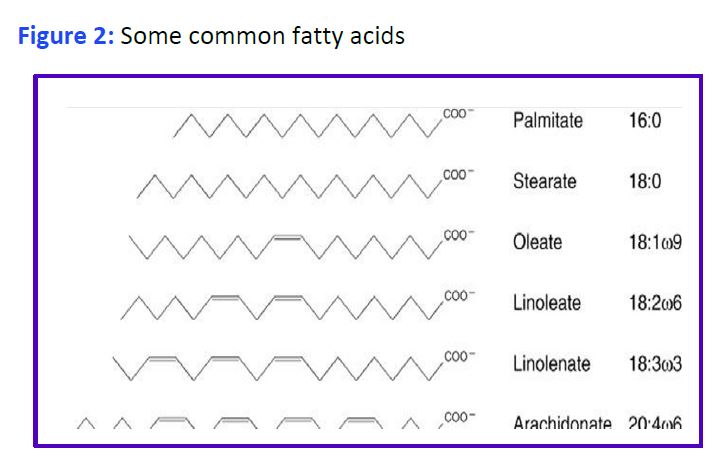
Chemical properties of Fatty Acids: Fatty acids consist of a long hydrocarbon chain and a hydrophilic carboxylic acid group. Systems of nomenclature for fatty acids are shown in Figure 1.

Free fatty acids (FFA or NE(non-esterified)FA) are poorly soluble in water. When present at too high levels their detergent properties cause disruption of cell membranes. FA circulate in plasma bound to the liver protein albumin. Various FA are components of many other molecules including glycerophospholipids, sphingolipids, cholesteryl esters and acylated proteins. The most abundant fatty acids have 16- or 18-carbon atoms although longer fatty acids (>20 carbons) are present in the lipids of most tissues. Double bonds, when present in a mammalian fatty acid, are always separated by a methylene (-CH2) and are of the cis configuration. Cis double bonds make the carbon chain less straight and cannot be packed as closely together (why butter (saturated) is solid while olive oil (unsaturated) is liquid at room temperature).
Structural representations of some common fatty acids are given in Fig. 2. Fatty acids that are linked to a hydrophilic phosphate containing group are called phospholipids, and are a main component of lipid bilayers. A fatty acid that is part of a different molecule is called an acyl group. Fatty acids are stored in the body as triglycerides primarily in adipocytes, but also in skeletal muscle and in liver. Excess TG storage in cells other than adipocytes eventually harms the cell.

Session Learning Objective 2 Part I. Diagram the pathways for fatty acid storage and release from adipose tissue and their regulation, highlighting the impact of insulin and counterregulatory hormones.
Mobilization and Utilization of fatty acids for energy
Triglycerides are stored in adipose tissue as large fat droplets within adipocytes. In order to be used as fuels, the fatty acids must be released and sent to tissues that require energy (such as liver or exercising muscle), transported, and oxidized. The products of complete oxidation of a fatty acid are CO2 and H2O.
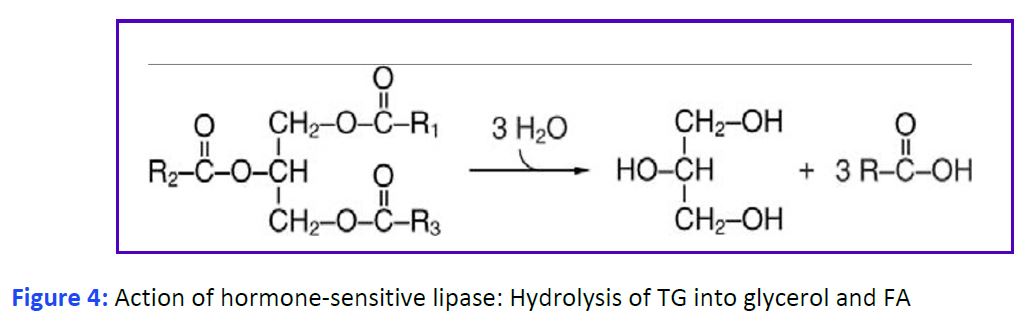
Release of fatty acids from triglycerides in white adipose tissue. Release of fatty acids involves the enzymatic hydrolysis of the ester bonds that link them to glycerol by one of several triglyceride lipases (Fig. 4). A major enzyme is hormone- sensitive lipase (HSL). The activity of this enzyme is controlled by phosphorylation through a G protein-coupled receptor (GPCR) cascade (similar to the activation of glycogen phosphorylase in carbohydrate metabolism). The hormone that consistently stimulates lipolysis by activating HSL is epinephrine signaling through β receptors. The roles of glucagon and growth hormone on lipolysis are less clear. The ability of cortisol to stimulate lipolysis vs lipid storage appears to be fat depot specific. The absence of insulin alone also activates HSL. Both fatty acids and glycerol are released from adipocytes into the blood. The free fatty acids immediately associate with plasma albumin. This increases their solubility and limits their deleterious detergent properties.
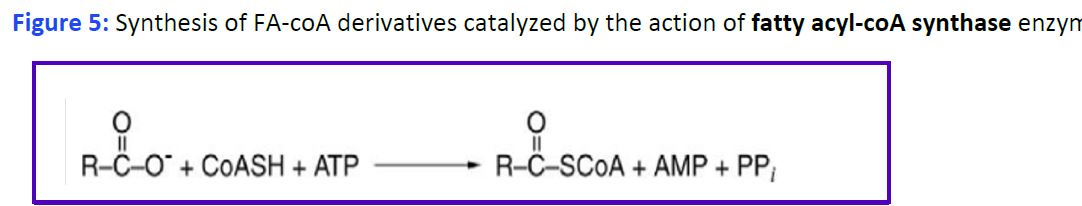
Uptake and activation of fatty acids and formation of Fatty Acyl-CoA. Fatty acids are taken up by various cells from the blood and used as fuels for metabolic processes. Uptake of fatty acids by cells is mediated by cellular transporters, and also involves cytosolic fatty acid-binding proteins, which act as intracellular carriers. Specific fatty acids can also bind to receptors (both membrane and cytoplasmic) and alter intracellular signaling and/or nuclear gene transcription. In the endoplasmic reticulum or the outer mitochondrial membrane fatty acids are activated by addition of coenzyme A (CoA), generating fatty acyl-coA derivatives. This reaction requires ATP and is mediated by fatty acyl-CoA synthetase (Fig. 5).
Entry of fatty acids into the mitochondrion. Fatty acid oxidation occurs in the mitochondrial matrix. Acyl-CoA molecules need a transport mechanism into the mitochondria because the inner mitochondrial membrane is not permeable to activated or free long chain fatty acids. The process involves a co-transport system in which a fatty acylcarnitine enters simultaneously with the exit of carnitine (Fig. 6). Synthesis of acylcarnitine in the intermembrane space is catalyzed by carnitine palmitoyltransferase-I (CPT-I), whereas regeneration of acyl CoA on the matrix side of the inner membrane involves an enzyme called CPT-II. Carnitine:acylcarnitine translocase facilitates the exchange of acylcarnitine and carnitine across the inner membrane to complete the process. An important control point is that CPT-I is inhibited by malonyl CoA, an intermediate in fatty acid synthesis (so that fatty acid synthesis and beta oxidation are not occurring in the same cell at the same time).

Board Focus: Extremely rare disorders of fatty acid mitochondrial transport are a board favorite. Inherited disorders due to CPT-I and CPT-II are described. The key to answering the question will be in the stem to determine where the acyl-CoA is getting stuck. Carnitine deficiency also occurs and can be inherited or acquired. This causes reduced fatty acid transport into the mitochondria. Critical illness is associated with acquired carnitine deficiency and you will commonly see ICU patients receiving carnitine supplementation.
Reactions of fatty acid oxidation. Four different reactions are used to oxidize fatty acids by 2C at a time into acetyl CoA and a shorter acyl CoA (Fig. 7). You will only be responsible for remembering Acyl CoA Dehydrogenase.
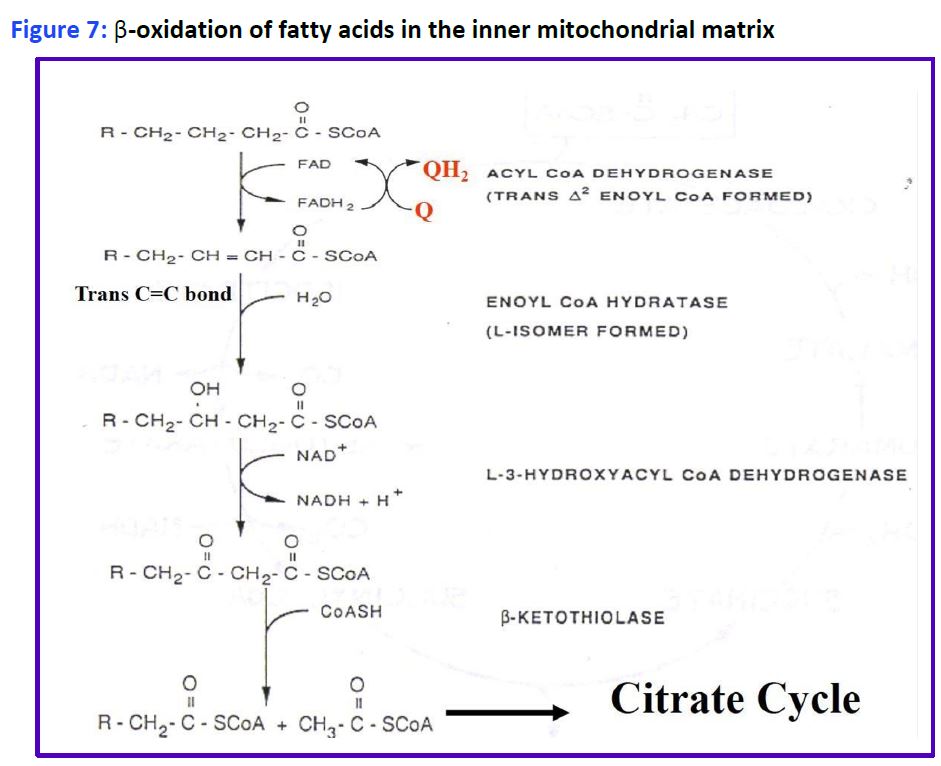
Figure 7: β-oxidation of fatty acids in the inner mitochondrial matrix.
The process is called β-oxidation, because it is the second or β-carbon that is cleaved from the fatty acid. Importantly, the oxidation reactions alone generate FADH2 and NADH in the mitochondrial matrix which can directly enter the electron transport chain to generate ATP. Importantly, this ATP production is not dependent on the TCA cycle and occurs even when the TCA cycle is inactive (eg. during gluconeogenesis). The acetyl CoA enters the TCA cycle as you have already seen in carbohydrate metabolism to generate even more ATP, or is packaged into ketones and released (see below). Additional rounds of acyl CoA oxidation and cleavage continue until the fatty acid is converted completely to acetyl CoA, if the fatty acid has an even number of carbons, eg: C16 palmitoyl CoA. Fatty acids with an odd number of carbon-atoms finish mitochondrial beta oxidation with the 3C propionyl-CoA which is rapidly converted to succinyl-CoA (4C intermediate) by enzymes used in amino acid catabolism and replenishes the TCA cycle (see Fig. 8).
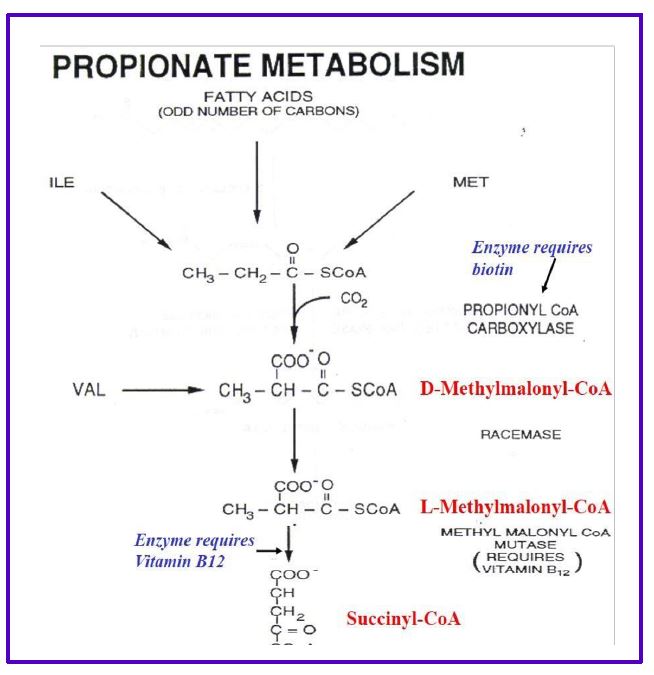
Figure 8. Propionate metabolism.
Fatty acid oxidation in the peroxisome. Peroxisomes take on some of the work of oxidizing fatty acids. These organelles have similar enzymes to oxidize fatty acids, but expressed from different genes. Peroxisomes mostly help metabolize fatty acids that are unsaturated, are longer than 22C, are branched or have an odd number of carbons. Additional enzymatic steps are needed to metabolize unsaturated fatty acids (needs a reductase) and branched chain fatty acids. The FADH2/NADH, propionyl CoA and Acetyl CoA generated in the peroxisome needs to be transferred to the mitochondria to be fully metabolized into ATP.
Practice questions:
1. How is fatty acid β-oxidation regulated?
2. What is the function of fatty acyl CoA synthase?
3. Why does the degradation of fatty acids yield more energy than the degradation of glucose?
4. Explain how fatty acids are transported across the inner mitochondrial membrane
5. What are the end products of fatty acid β-oxidation?
SLO3. Outline the pathway and regulation of de novo lipid biosynthesis.
Fatty Acid Biosynthesis
Goals: 1. Define the similarities and differences between fatty acid synthesis and fatty acid oxidation; 2. Understand how fatty acid synthesis is regulated, and how this differs from the pathway of fatty acid degradation.
Cytoplasmic fatty acid synthesis: Fatty acids are synthesized by stepwise addition of 2-carbon units to the carboxyl end of acetate. The 2- carbon units are derived from malonyl CoA, a 3-carbon compound, generated from Acetyl-CoA and CO2. This reaction is catalyzed by acetyl CoA carboxylase (ACC) and occurs in the cytoplasm.
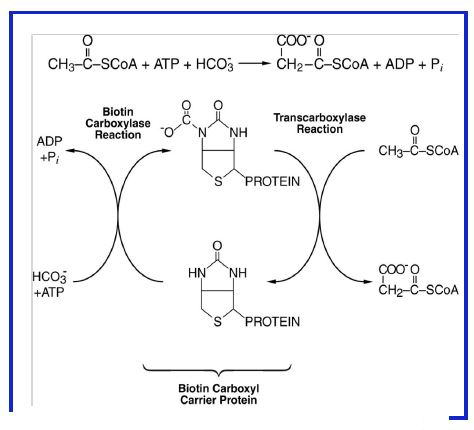
Figure 9: Synthesis of malonyl-CoA from acetyl-CoA, CO2/HCO3-, and ATP by Acetyl- CoA Carboxylase.
Synthesis of Malonyl-CoA. This reaction occurs in two steps (Fig. 9) and involves a multi-enzyme complex. ATP is required to generate an activated intermediate (carboxybiotin) followed by transfer of the CO2 group to acetyl CoA. This reaction is the rate limiting step of fatty acid synthesis and is the main site of control of the pathway.
Session Learning Objective 2 Part II. Diagram the pathways for fatty acid storage and release from adipose tissue and their regulation, highlighting the impact of insulin and counterregulatory hormones.
Regulation of ACC
Short term regulation of ACC: Cytoplasmic citrate is an allosteric activator (positive feedback). The enzyme is inactivated by palmitate, the end product of the fatty acid synthase (FAS) reaction (negative feedback). The activity of ACC is also controlled by hormones; epinephrine and glucagon indirectly stimulate phosphorylation of ACC, which inactivates the enzyme, while insulin indirectly causes dephosphorylation and activates the enzyme (similar to how the activity of glycogen synthase is regulated by these hormones).
Long term regulation of ACC: The ACC enzyme is under complex transcriptional control affecting levels in the cell, including by insulin and glucagon. A high carbohydrate and low fat diet also increases ACC gene transcription.

Figure 10: Allosteric and hormonal regulation of acetyl coA carboxylase.
Reactions catalyzed by fatty acid synthase (FAS). FAS consists of two copies of a large protein which has all the activities needed for synthesis of palmitate. Processing of the growing fatty acid by the dimeric enzyme occurs while it remains covalently attached to the enzyme. During biosynthesis, the elongating fatty acid chain and malonyl-CoA are covalently bound to FAS via two different sulfhydryl groups (Figure 11), including one originating from a coenzyme-A like moiety referred to as “acyl carrier protein” (ACP). ACP tagging of fatty acids is a characteristic feature of FA synthesis, in contrast to -CoA tagging for degradation. The details of Fatty acid Synthesis are depicted below, but we only expect you to be aware of Acetyl CoA Carboxylase and its regulation as the rate limiting step in fatty acid synthesis (outlined above).

Figure 11.
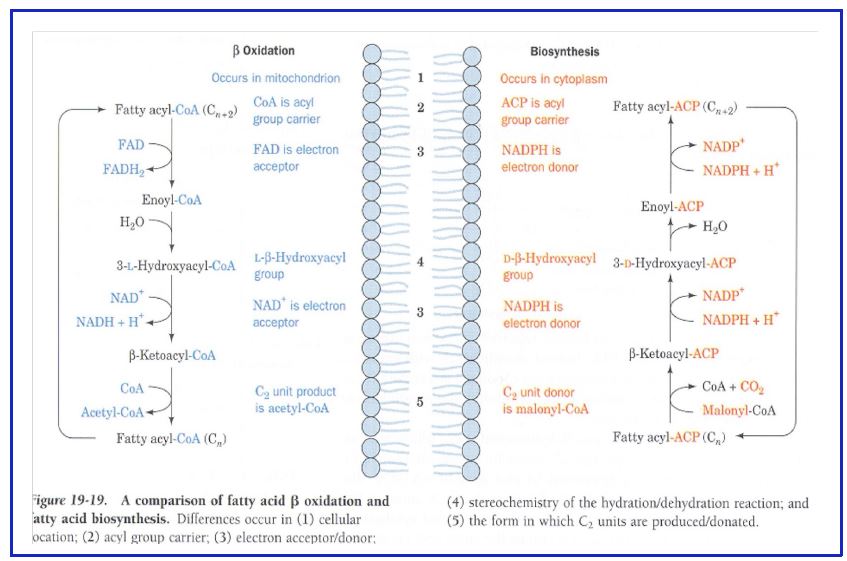
Figure 12: Comparison between fatty acid synthesis and fatty acid oxidation
Transport of acetyl-coA to the cytoplasm for fatty acid synthesis. The coenzyme-A portion of acetyl-CoA cannot cross the inner mitochondrial membrane. Thus, in order for acetyl-coA to be transported from the mitochondrial matrix to the cytosolic side of the inner mitochondrial membrane, acetyl-coA must first condense with oxaloacetate (OAA, also can’t cross the inner mitochondrial membrane) to form citrate. Citrate is then transported across to the cytosolic side, where it is re-converted to OAA and acetyl-coA by the action of ATP citrate lyase. Cytosolic OAA is reduced back to malate, which undergoes oxidative decarboxylation by action of the malic enzyme to generate NADPH, CO2, and pyruvate. The NADPH produced is used as reducing power for fatty acid synthesis in the cytosol; pyruvate is transported back to the mitochondrial matrix to be reconverted to OAA by the action of pyruvate carboxylase (Figure 13). It is important to regenerate OAA, as it is a key intermediate of the TCA/Krebs cycle.

Figure 13: Transport of acetyl-CoA from the mitochondrial matrix to the cytoplasm.
Elongation and Desaturation: The product of FAS (palmitate) has 16 carbons and no double bonds. Palmitate can be elongated and desaturated to produce long chain fatty acids with multiple double bonds. Elongation to 18, 20 and 22 carbon fatty acids occurs in two compartments: the mitochondrion where acetyl CoA is used for elongation, and on the membranes of the endoplasmic reticulum where malonyl-CoA is used. The additional 2-carbon units are added to the carboxyl end of the fatty acid in both cases.
Introduction of double bonds (desaturation): Most organisms possess fatty acyl CoA desaturases that introduce double bonds into fatty acids. Remember the double bonds are always in the cis-configuration. Most of you have heard of omega-3 fatty acids (from fish oils). This is an older nomenclature and indicates where the first double is located counting from the last carbon (not affected by elongation). A more modern system uses a delta (Δ) and a superscripted number to locate the position of the double bond relative to the carboxyl of the fatty acid (see Fig. 1). Humans have three fatty acyl CoA desaturases with specificities for introduction of a double bond at Δ – 9, Δ -6, or Δ -5 positions. Synthesis of fatty acids generally follows these rules:
1. The first double bond is introduced between carbons 9 and 10 (Δ – 9 position), counting from the carboxyl.
2. If a double bond is already present, the next one is introduced closer to the carboxyl, separated from the first by a methylene (-CH2).
Thus, combinations of desaturation and elongation can lead to synthesis of a variety of unsaturated fatty acids (Fig. 14).
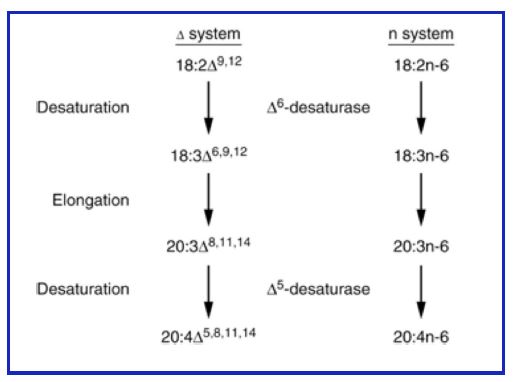
Figure 14: Elongation and desaturation of fatty acids.
However, humans are unable to synthesize fatty acids with double bonds farther from the carboxyl end, eg. Δ12 or Δ15. These fatty acids are considered ‘essential fatty acids’ because they need to be consumed in the diet to prevent pathology. The main essential fatty acids are linoleic acid (18:3), eicosapentaenoic acid (20:5) and docosahexaenoic acid (22:5). All of these fatty acids have double bonds at distant positions (omega 3 or omega 6).
Practice questions:
1. How is fatty acid synthesis regulated?
2. What is the function of acetyl-coA carboxylase (ACC)?
3. How is the activity of the ACC enzyme regulated?
4. How does fatty acid synthesis differ from beta oxidation of fatty acid
5. Explain how acetyl coA is transported from the mitochondrial matrix and across the inner mitochondrial membrane to the cytosol to be made available for fatty acid synthesis.
6. How are fatty acids longer than 16-carbon chains produced?
7. Now that you understand the role of citrate in catabolism and anabolism, can you explain why citrate inhibits PFK-1, the key committed step of glycolysis?
Session Learning Objective 4. Explain how and why ketone bodies are formed and how they are utilized.
Ketone Synthesis and Metabolism
Goals. 1 Understand where, when and why ketone bodies are synthesized. Identify which tissues can metabolize ketones for fuel. 3. Describe the adaptations that need to occur for neurons to metabolize ketones.
Ketone production and utilization. Fatty acid mobilization and metabolism doesn’t always lead to complete oxidation to CO2 in the liver. Acetoacetate and β-hydroxybutyrate, commonly referred to as ketones (also called ketone bodies) are also produced from fatty acids and exported to other tissues (heart, skeletal muscle, others) where they are used as sources of acetyl CoA for energy production. Ketone synthesis (Fig. 15) starts with a thiolase joining two acetyl CoA molecules to form acetoacetyl CoA. Then HMG-CoA synthase (mitochondrial) adds another acetyl-CoA to produce β-hydroxymethylglutaryl CoA (HMG-CoA). This product is cleaved to yield acetyl CoA and acetoacetate by HMG-CoA lyase. β-hydroxybutyrate is produced from acetoacetate by a dehydrogenase requiring NADH. The liver has the largest amounts of HMG-CoA lyase (the cleavage enzyme) and hence is the main site of acetoacetate production. Note the HMG-CoA in the mitochondria is different isozyme from and regulated differently from HMG-CoA in the cytoplasm which is essential for cholesterol synthesis.
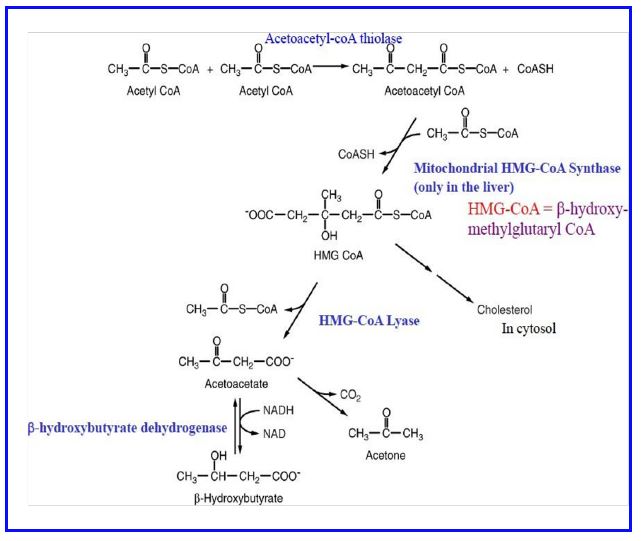
Figure 15: Biosynthesis of ketones (in liver mitochondria).
Ketones are readily taken up from the blood by many tissues via a family of monocarboxylate transporters (MCT). The brain normally does not metabolize ketones for fuel. With prolonged (48-72 hour) starvation both the MCTs and the mitochondrial enzymes are upregulated and neurons begin to use ketones efficiently for fuel. This adaptation by neurons is a critical step in reducing the amount of glucose the liver (and kidneys) need to produce through gluconeogenesis. Metabolism of β-hydroxybutyrate occurs in mitochondria and involves oxidation to acetoacetate, activation by conversion to acetoacetyl CoA, cleavage to 2 molecules of acetyl CoA by β-ketothiolase, and oxidation of acetyl CoA via the TCA cycle. Activation of acetoacetate utilizes succinyl CoA and an enzyme that catalyzes an acyl- transfer reaction (SCOT, succinyl CoA ketoacid CoA transferase) (Figure 16). Non-neuronal cells in the brain, astrocytes being the best example, can metabolize fatty acids (neurons cannot) and can produce ketones in low amounts for neurons to metabolize.
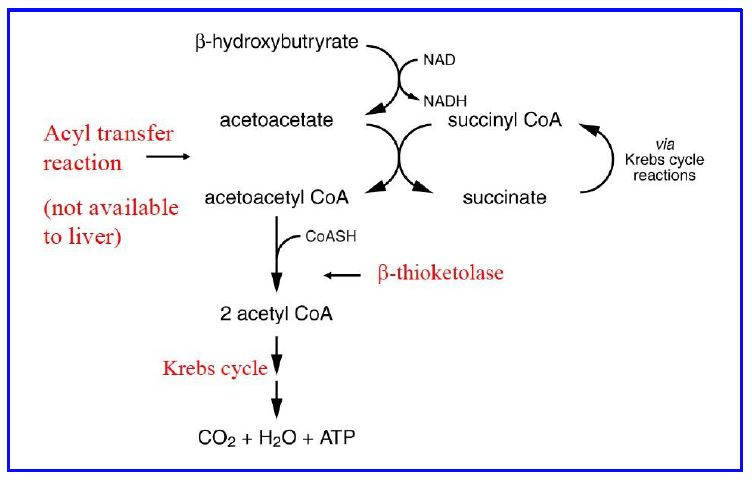
Figure 16: Metabolism of β-hydroxybutyrate in tissues (activation & oxidation of ketones for energy production.
The liver is the most active organ in the production of acetoacetate but lacks the acyl transferase necessary to metabolize ketones, thus ensuring that hepatic ketones exit the cell and are not metabolized by hepatocytes. Ketone metabolism is important in many tissues during fasting as a fuel source. Ketones can accumulate in the blood in various conditions. The most common is Type 1 Diabetes, when very low insulin signaling causes very high rates of triglyceride lipolysis and very high rates of ketone production. This can lead to diabetic ketoacidosis. Other conditions (such as ethanol toxicity) can also cause ketoacidosis. Fig. 16 summarizes the mobilization of triglycerides and subsequent metabolic processing of fatty acids for energy or ketone synthesis.

Figure 17: Summary of fatty acid oxidation and ketone body production and utilization.
Session Learning Objective 5. Understand the biochemical mechanism behind examples of lipid metabolism dysfunction, including carnitine deficiency and medium chain fatty acyl CoA deficiency.
Boards Focus: Infants presenting with hypoglycemia and lethargy (failure to thrive) is a common clinical scenario on the boards (not common in practice). Always check the question stem to see if ketones are present. If ketones are present then lipid oxidation is occurring and the diagnosis is likely related to carbohydrate metabolism. If ketones are absent then the problem is with fatty acid transport or fatty acid oxidation (see below).
Inherited diseases of fatty acid oxidation. Several different fatty acyl CoA dehydrogenases catalyze the first step of fatty acid oxidation, including those specific for very long chain fatty acids (VLCAD, very long chain acyl CoA dehydrogenase), long-chain (LCAD), medium chain (MCAD) and short chain (SCAD). Rare genetic disorders cause disorders related to the function of these enzymes. Mutations are most common in the gene for MCAD and result in fasting hypoglycemia. This secondary disturbance in carbohydrate metabolism resulting from a primary defect in the ability to efficiently oxidize fatty acids points out the extent to which fuel metabolic pathways are related to one another. Gluconeogenesis is required for plasma glucose homeostasis during fasting and requires considerable energy (as we have seen earlier). The inability to oxidize fatty acids efficiently reduces the amount of energy that can be derived from fat metabolism and the diminished gluconeogenesis results in fasting hypoglycemia.
Practice questions:
1. Why are ketone bodies only made in the liver?
2. What are ketone bodies used for in peripheral tissues?
3. Why is ketone body metabolism so relevant to Type 1 diabetes and fasting?
4. Explain how ketone bodies are made.
5. Explain how ketone bodies are activated and oxidized in tissues such as the CNS.
6. In which organelle are ketone bodies synthesized?
Session Learning Objective 6. Compare the significance of arachidonate derivatives (COX inhibition by aspirin, prostaglandins, leukotrienes).
Session Learning Objective 7. Recognize the roles of eicosanoids in diverse physiological processes and their roles as inflammatory mediators.
Arachidonic Acid and Eicosanoid Production.
Goals: 1. Understand the roles of eicosanoids in diverse physiological processes 2. Appreciate the tissue specific pathways for eicosanoid biosynthesis.
Eicosanoids are cellular signaling molecules. These molecules give cells the opportunity to change their own activity (autocrine) or alter the activity of surrounding cells (paracrine), but usually do not cause a signal in distant tissues (endocrine). While endocrine mediators (hormones) such as insulin are produced in large quantities by specialized glands, eicosanoids can be produced in very small amounts in almost all tissues. Eicosanoids are not stored and have a very short half life, being rapidly metabolized to inactive products. Most eicosanoid signals are produced as part of a cellular response to stress. Effects include a wide range range of responses, both physiological/tissue protection and repair, and pathological/hypersensitivity and allergy responses. Eicosanoids can cause the classic four signs of inflammation, pain (dolor), warmth (calor), redness (rubor) and swelling (tumor), through vasodilation, changes in vascular permeability and triggering pain sensors. As key mediators of inflammation, there are multiple medications that target eicosanoid pathways.

Figure 18. Arachidonic Acid and phospholipase A2.
Release of arachidonic acid. Arachidonic acid is a 20 carbon fatty acid and a precursor for all major classes of eicosanoids, but the free concentration of this fatty acid in cells is very low. Eicosanoid production begins with release of arachidonic acid (Fig. 17). A very specific lipase called phospholipase A2 catalyzes the release of arachidonic acid from the middle carbon (sn2 position) of a glycerophospholipid (often phosphotidylcholine) in the inner leaflet of a lipid bilayer. Once the fatty acid enters is released, the metabolic pathway that arachidonic acid enters and the eicosanoid end products depend on tissue and cell type (Figure 18). With the exception of red blood cells, nearly all human cells can generate eicosanoid signals. Arachidonic acid is not the only fatty acid used for generating eicosanoids, but is the major metabolite.
Modification of arachidonic acid. We focus on two major pathways that modify arachidonic acid into eicosanoid signals. These are the cyclooxygenase and lipoxygenase pathways.
1. Cyclooxygenase pathway gives rise to the:
• Prostaglandins
• Thromboxanes
2. Lipoxygenase pathway gives rise to the:
• Leukotrienes
• Hydroxylated eicosanoids
• HETE = hydroxyeicosatetraenoic acid; diHETE = dihydroxyeicosatetraenoic acid. Most common: 20-HETE – hydroxyl on C-20 of arachidonate; stimulates vascular constriction (vasoconstriction), except in the lungs where 20-HETE stimulates pulmonary vasodilation.

Figure 19: The eicosanoid biosynthesis pathway.
The cyclooxygenase pathway.
After release from a glycerophospholipid, arachidonate can interact with a
membrane protein, PGH2 synthase, prostaglandin H2 synthase, commonly known as cyclooxygenase (COX).
Cyclooxygenase (COX) has two distinct active sites to catalyze the conversion of arachidonate to PGH2 (Fig. 19). The first activity which adds oxygen to arachidonate is the cyclooxygenase reaction (Fig 19). The second activity of the enzyme requires NADPH to provide reducing power.
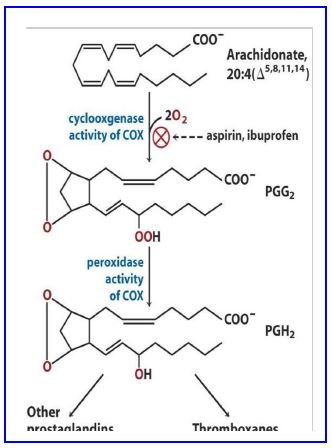
Figure 20. Modifications by cyclooxygenase.
COX isoforms. There are two main COX isoforms. COX-1 is constitutively expressed in most tissues at low levels and is important for key maintenance functions in epithelial tissue including the gut mucosa and the kidney. The expression of COX-2 is more restricted and is induced by inflammatory signals. COX-1 and COX–2 are coded by separate genes.
Tissue specific production of prostaglandins. Most tissues produce a relatively limited set of eicosanoids from arachidonate because of the tissue-specific expression of biosynthetic enzymes. For instance, PGI2 (prostacyclin) promotes vasodilation and decreases platelet aggregation and is a major product of vascular endothelium (in contrast thromboxane A2 increases platelet aggregation and is produced predominantly by activated platelets). PGD is a bronchoconstrictor and is a major product of mast cells.
Modes of action of prostaglandins. In general, eicosanoids are secreted from cells and act on the parent cell (autocrine action) or adjacent cells (paracrine action). Their instability and secretion in low amounts limits the effective range of their action. Prostaglandins exert their effects on cells through interaction with GPCRs (G protein-coupled receptors) affecting the activity of adenylate cyclase or PLC (phospholipase C). Those receptors coupled to Gs (stimulatory G-protein) stimulate adenylate cyclase which increases the concentration of cAMP. Those coupled to Gi (inhibitory G-protein) inhibit adenylate cyclase, decrease cAMP concentration, and are antagonistic to the Gs group. Those coupled to Gq stimulate PLC increase IP3 (inositol trisphosphate), which increases Ca2+ concentration. Nine classes of G-protein coupled receptors (GPCRs) have been discovered that mediate the effects of eicosanoids.

Figure 21. Prostaglandin and Thromboxane Summary.
Physiological roles of prostaglandins. This block will not cover a definitive list of all the physiological processes affected by prostaglandins because their effects are so diverse. The following section touches only on select, well-defined physiological roles.
PGE2; resorption of sodium in the kidney glomerulus; pain pathways via sensitization of neurons; house-keeping functions in gastric mucosa.
PGI2 and TXA2: the opposing roles of these PGs in vascular health has been mentioned above
PGF2α: contraction of uterine smooth muscle
PGD2: release of histamines from sensitized mast cells, vasodilator, bronchoconstrictor.
Pharmacology associated with prostaglandins. Several different classes of drugs affect prostaglandin biosynthesis and/or molecular action.
Glucocorticoids. Prednisone and dexamethasone, are examples of synthetic glucocorticoids and are frequently prescribed for relief of symptoms from inflammatory diseases. One anti-inflammatory effect of glucocorticoids is the inhibition of cytoplasmic phospholipase A1 which decreases the production of eicosanoids by preventing arachidonic acid release from the lipid bilayer.
Glucocorticoids have a wide variety of other anti-inflammatory effects on cytokine (more complex signaling molecules you will hear about later) production and gene expression of pro-inflammatory mediators in many tissues.
Aspirin. Willowbark is a traditional medicine used to alleviate fever and pain. This knowledge led to the discovery of acetyl salicylic acid (ASA or aspirin) as a pain reliever. Aspirin inhibits the activities of COX-1 and COX-2 through irreversible covalent acetylation of the enzyme, which blocks the first active site.
NSAIDs (non-steroidal anti-inflammatory drugs). A number of inhibitors of COXs have been developed by pharmaceutical companies, which collectively are known as NSAIDs to differentiate them from the steroid (glucocorticoid) analogues mentioned above. Examples of NSAIDs include ibuprofen and naproxen. These first generation drugs are relatively non-specific and inhibit both COX-1 and COX-2 to approximately the same extent. Their anti- inflammatory properties are thought to result from inhibition of COX-2 whereas their side effects (gastrointestinal bleeding, prolonged clotting times, renal failure, etc.) are believed to result from inhibition of COX-1. Ibuprofen and NSAIDs other than aspirin bind tightly, but non-covalently to COXs.
COX-2 inhibitors. Specific COX-2 inhibitors were developed in an effort to reduce the side effects associated with use of NSAIDs by not inhibiting COX-1. The medications worked in reducing joint pain without causing stomach or kidney side effects, but two medications were withdrawn from the market because long term studies showed they increased risk of heart attack (myocardial infarction) and stroke (cerebrovascular disease). One selective COX-2 inhibitor with a much shorter half life is still on the market. Why did COX-2 inhibitors cause cardiovascular disease? The vascular endothelial product PGI2 decreases cardiovascular disease by reducing platelet aggregation, increasing vasodilation, and decreasing proliferation of vascular smooth muscle cells (SMC). In contrast, TXA2 increases platelet aggregation, promotes vasoconstriction, and increases proliferation of SMC. Thus, vessel health is highly dependent on balanced production of these two antagonistic mediators. Aspirin use is associated with reduced risk of myocardial infarctions and embolic stroke because the drug irreversibly inhibits COXs in both platelets and vascular endothelial cells. Endothelial cells, however, can rapidly renew COX levels via protein turnover (transcription and translation) whereas platelets are anuclear and are unable to synthesize new COX. Thus, the PGI2 /TXA2 balance is tipped in favor of PGI2. COX-2 is responsible for PGI2 production in vascular endothelium whereas TXA2 synthesis in platelets is largely via COX 1. Thus, specific COX-2 inhibitors with a long half life might be expected to tip the balance in favor of TXA2 and increase the risk of cardiovascular disease.
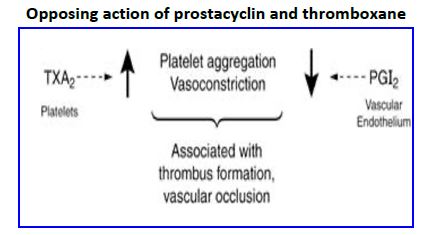
Figure 22. Opposing action of prostacyclin and thromboxane.
Acetaminophen (Tylenol):
This medications is an NSAID that decreases fever and pain through interaction with the endogenous cannabinoid system and does not have significant direct effects on eicosanoids
The Lipoxygenase Pathway:
There are six human lipoxygenase enzymes that are expressed in different tissues and at different levels. Sometimes two lipoxygenases act sequentially on arachidonic acid to generate the final eicosanoid product. Lipoxygenase inhibitors are currently less commonly used than cyclooxygenase inhibitors. A pharmacological lipoxygenase inhibitor is available for the treatment of chronic asthma.
LEUKOTRIENES:
Leukotrienes constitute the second major class of compounds derived from arachidonate. In contrast to the prostaglandins, they are all mediators of inflammation.
LIPOXINS:
These eicosanoids play important roles as anti-inflammatory lipid mediators important in resolution of inflammation and they and their receptors are important targets for pharmacological intervention in chronic inflammatory diseases.
Feedback: