
13 Amino Acid, Heme and Bilirubin Disorders
Amino Acid, Heme and Bilirubin Disorders
WWAMI FMR
Pamela Langer, PhD, WWAMI-WY
Session Learning Objectives
SLO 2: Relate the disruption of the heme biosynthetic pathway to porphyrias and lead poisoning.
SLO 3: Describe the importance of heme degradation in the development of hyperbilirubinemia.
Amino Acid, Heme, Bilirubin Overview
An imbalance in the amino acid pool, causing either excesses or deficiencies of amino acids, can result from genetic disorders, such as phenylketonuria (PKU) and homocystinuria. Other disorders addressed in this chapter are associated with either the synthesis or degradation of heme, the prosthetic group for hemeproteins such as hemoglobin. Note that there are many other genetic disorders related to amino acids and amino acid derivatives that are important but are not covered in this session only because of time constraints.
SLO 1: Discuss the enzymatic defects in, and clinical consequences of, phenylketonuria (PKU) and homocystinuria.
With genetic defects causing a pathological consequence, the severity of the disorder depends on the specific mutation. For example, a mutation that causes the complete absence of a protein (from a null mutation) is likely to cause a more severe phenotype than a missense mutation, especially one that causes a conservative amino acid substitution. Clinical symptoms in genetic disorders may be the result of a deficiency of an amino acid, an accumulation of substrates of a defective enzyme, or the utilization of minor metabolic pathways causing an abnormal excess of certain products.
Phenylketonuria (PKU)
Most cases of PKU result from autosomal recessive defects in the gene encoding phenylalanine hydroxylase (PAH), that catalyzes hydroxylation of phenylalanine (Phe) to tyrosine (Tyr). Over 1000 mutations have been identified in the PAH gene, and the worldwide prevalence of PAH is estimated at about 1/10,000 in populations with European ancestry. Milder, rarer forms of PKU occur if there is a deficiency in regeneration of the coenzyme for PAH, tetrahydrobiopterin (BH4), and treatment may be different than for PKU. PAH deficiency is associated with a range of intellectual disabilities depending on the severity of the defect and the effectiveness of treatment. Mechanisms underlying development of intellectual disabilities are incompletely characterized, but several potential mechanisms related to excess Phe have been proposed.
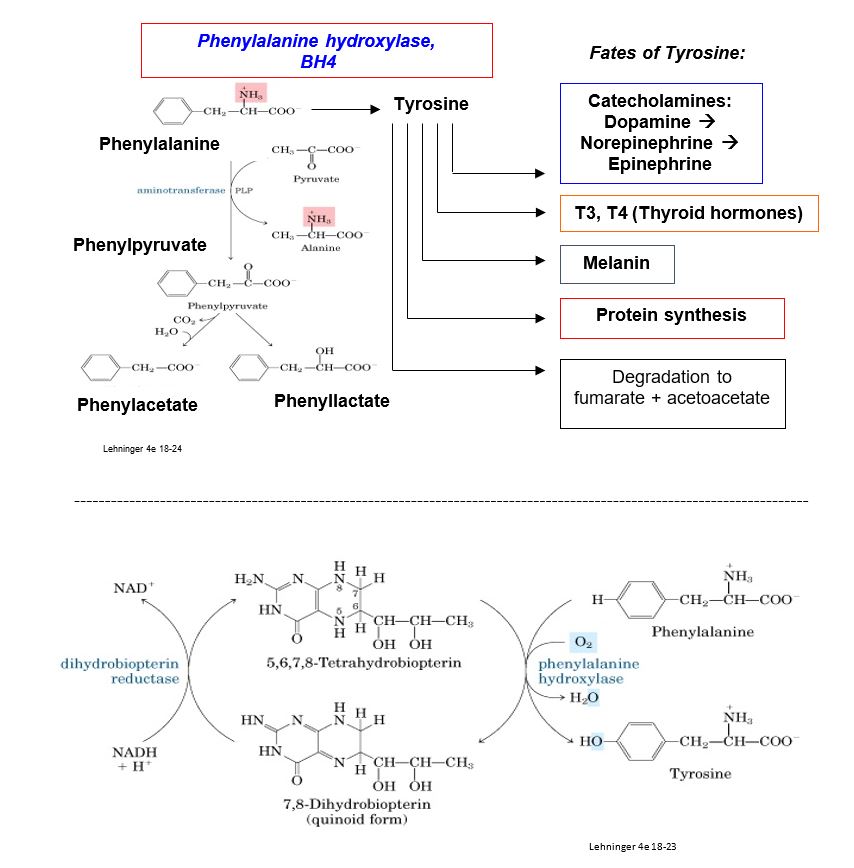
Figure 1.
Metabolic consequences of PKU include elevated blood and urine concentrations of Phe and its derivatives phenylacetate and phenyllactate. Although endogenous synthesis of tyrosine may be deficient, tyrosine is also obtained from the diet, and its physiological level may be normal or low-normal. Products synthesized from a tyrosine precursor are not necessarily deficient in PKU, e.g. hypothyroidism is a rare presentation for PKU. However, tyrosinase, an enzyme in the melanin biosynthetic pathway, is inhibited with high Phe. Consequently, individuals with PKU typically present with hair and skin with very little pigmentation.
There are many hypotheses for the connection between biochemical abnormalities and development of intellectual disability, but the mechanism is incompletely understood. One (controversial) hypothesis involves the Large Neutral Amino Acid (LNAA) transporter. This LNAA transporter is the only transporter for large neutral amino acids, such as Phe, Tyr and Trp, across the blood brain barrier. Inhibition of this transporter with elevated Phe in PKU may cause a deficiency in transport of Tyr and Trp across the blood brain barrier (precursors for catecholamines and serotonin, respectively.)
Although a person may become more tolerant of dietary Phe with increasing age, women with PKU are advised to follow a Phe-restricted diet for at least three months before becoming pregnant and throughout pregnancy. The synthetic sweetener Aspartame, aspartyl-phenylalanine methyl ester, should be avoided at all times.
If a mother with PKU on a Phe-unrestricted diet becomes pregnant, elevated serum Phe levels can cause phenylalanine embryopathy (maternal PKU syndrome) regardless of the genotype of the fetus. Phe levels are higher in the fetus than the mother and may reach a concentration that is teratogenic. However, the mechanism of the teratogenic potential of elevated Phe and/or the Phe derivatives phenylacetate and phenyllactate, is not known. Unmanaged maternal PKU syndrome is at high risk for birth of a baby with intellectual disability, microcephaly, congenital heart disease, and low birth weight. All newborns in the US should be screened for PKU as part of a newborn screening panel, as impaired brain development may be minimized in a newborn by initiating a low-Phe diet at the earliest possible time after birth and within the first week of life.
Although it is beyond the scope of this brief discussion of PKU, various treatment strategies for PKU have been developed, including administration with a synthetic BH4. The newest addition to the list is a phenylalanine-degrading enzyme called phenylalanine ammonia lyase, originally derived from a prokaryote and produced as a recombinant form conjugated with polyethylene glycol (PEG) to minimize immunogenicity. Pegvaliase, considered to be the first in its class as a new medication and approved by the FDA for use in 2018, has the goal of degrading Phe to lower the level of Phe in the blood. Unfortunately, severe allergic reactions are a common side effect of the drug, so the first dose must be administered in a healthcare setting in case of an anaphylactic response.
Homocystinuria
The predominant autosomal recessive defects associated with homocystinuria are found in the gene encoding cystathionine beta-synthase (CBS), a pyridoxal phosphate (PLP)-requiring enzyme in the cysteine biosynthetic pathway. The biochemical consequence of this deficiency is an increase in homocysteine (Hcys) and its disulfide-bonded form, homocystine. The side chain of homocysteine (-CH2-CH2-SH) is longer than that of cysteine (-CH2-SH). Homocysteine is an essential part of the folate and S-adenosylmethionine (SAM) cycles and is a substrate for the shared reaction catalyzed by methionine synthase at the intersection of these two cycles.
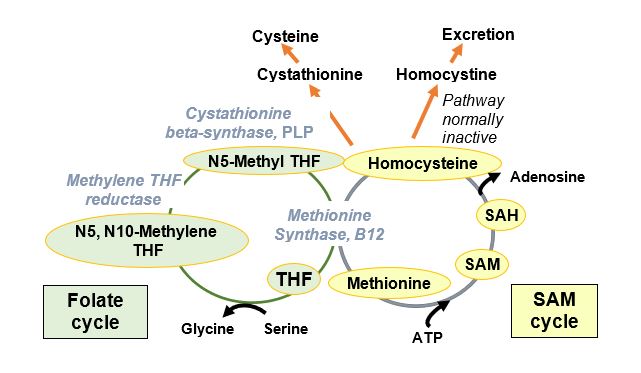
Figure 2
Worldwide prevalence of homocystinuria is reported as 1/200,000 to 1/335,000, but higher in certain populations (Ireland, Germany, Norway, Qatar). Patients develop hyperhomocystinuria, hyperhomocysteinemia, cysteine deficiency, and physical abnormalities that could result from a number of biochemical consequences. One of the biochemical consequences of elevated Hcys is the inhibition of production or activity of lysyl oxidase (LOX), an enzyme important in collagen cross-linking. Elevated Hcys may also promote the SAM cycle and thus methylation affecting transcription.
A newborn may first appear normal but later present with symptoms including developmental delay, Marfanoid appearance, osteoporosis, ocular abnormalities, thromboembolic disease, severe premature atherosclerosis and extreme elevations in plasma Hcys. Elevated Hcys is considered an independent risk factor for increased risk of coronary heart disease (CHD).
SLO 2: Relate the disruption of the heme biosynthetic pathway to porphyrias and lead poisoning.
Heme is the prosthetic group for diverse hemeproteins, including hemoglobin, myoglobin, cytochrome P450 enzymes, cytochrome c in the electron transport chain (and apoptosis), catalase acting as an anti-oxidant catalyzing the degradation of hydrogen peroxide (H2O2) to water, and thyroid peroxidase, essential in thyroid hormone biosynthesis.
Starting with the amino acid glycine and succinyl CoA, heme is synthesized via the porphyrin biosynthetic pathway, a multi-step pathway distributed between the mitochondrion and cytosol and operating in most cells (see figure for comments by enzyme numbers). ALA synthase (5-aminolevulinic acid synthase-1, ALAS-1), the first enzyme of the porphyrin pathway, is the rate-controlling step. ALAS-1 is highly inducible and responds to changes in nutritional status (e.g. glucose levels) and use of certain drugs. Normally, heme causes feedback inhibition of ALA synthase. When the porphyrin pathway is compromised, porphyrin pathway intermediates are elevated, and oxidized colored porphyrin intermediates can be detected clinically.
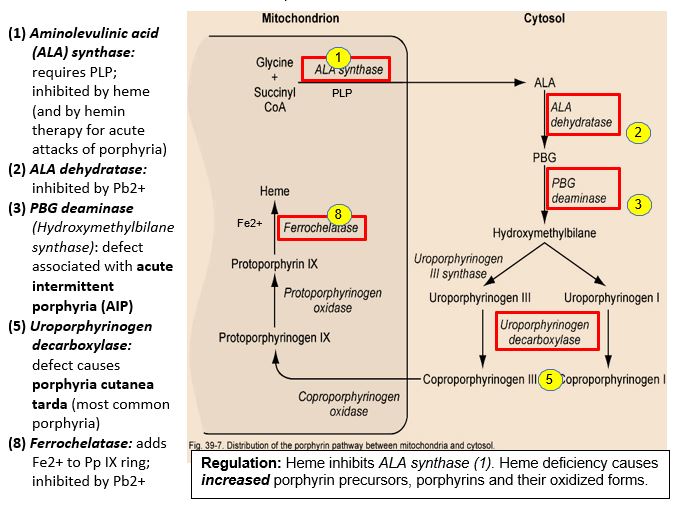
Figure 3: Regulation: Heme inhibits ALA synthase (1). Heme deficiency causes increased porphyrin precursors, porphyrins and their oxidized forms.
Enzyme defects in porphyrin biosynthesis or inhibition of the pathway can lead to heme deficiency and neurological/neuromuscular and cutaneous porphyrias. Secondary disorders of porphyrin metabolism are caused by lead poisoning or iron deficiency.
Neurological porphyria, such as an acute hepatic porphyria (e.g. acute intermittent porphyria, AIP) may present with symptoms that include abdominal pain and neuromuscular and psychiatric disturbances. Photosensitization in cutaneous porphyrias results from accumulated, photo-excited porphyrins that are reactive. Cutaneous porphyrias are blistering cutaneous porphyria (e.g. porphyria cutanea tarda, PCT, the most common porphyria) and acute nonblistering cutaneous porphyria. Genetic testing is typically used in diagnosis of porphyrias.
Lead (Pb) poisoning
Lead (Pb2+) inhibits two enzymes in the porphyrin pathway: ALA dehydratase, the second step, and ferrochelatase, catalyzing the addition of Fe2+ to protoporphyin IX. Lead is a neurotoxin that accumulates in soft tissues and bones by binding to sulfhydryl groups, normally bound by Ca2+, Fe2+, Zn2+. It has been observed that Pb2+ concentrated in bones may be released later in life as bone is remodeled. Symptoms of lead poisoning include irritability, loss of appetite, weight loss, lethargy, abdominal pain, vomiting, constipation, and learning difficulties. Although the nervous system and kidney are more commonly affected, anemia may result, but only with high lead levels. In treating lead poisoning, it is imperative to remove the source, which is often chips of paint containing lead. It may be more practical, economical and safer to paint over old paint rather than try to remove it. With high dose lead exposure, a chelating agent such as EDTA (ethylenediaminetetraacetic acid) is used to sequester divalent Pb2+ cations.
Acute intermittent porphyria (AIP)
AIP is the most common acute hepatic porphyria, with a prevalence of approximately 5/100,000 in the US, including individuals who are pre-symptomatic, and a higher prevalence in Sweden, because of a founder effect. AIP is caused by an autosomal dominant defect in the HMBS gene encoding Porphobilinogen (PBG) deaminase (also called Hydroxymethylbilane (HMB) synthase). The genetic deficiency alone is not sufficient to cause a clinical presentation, and there is a low penetrance of the genetic disorder. AIP rarely presents before puberty and often remains in a latent phase such that many patients never have symptoms.
The first sign of an AIP attack is often an intense, diffuse abdominal pain that is constant. Some people have psychological manifestations including insomnia, anxiety, depression, hallucinations and other states of altered consciousness. AIP attacks range from mild to potentially life-threatening acute attacks that include severe abdominal pain accompanied by nausea, vomiting, tachycardia and hypertension.
Although AIP is an autosomal dominant disorder, presentation of AIP is intermittent in that symptoms may occur only after a patient with AIP experiences stresses such as inadequate caloric intake (e.g. with fasting or heavy exercise), excess alcohol intake, menstrual hormonal changes, illness, surgery, or if they are exposed to substances that stimulate porphyrin biosynthesis or the cytochrome P450 enzymes.
Certain prescribed and illicit drugs detoxified by hepatic cytochrome P450 enzymes, or drugs that induce ALA synthase-1 expression, may also induce an attack, e.g. barbiturates, anti-epileptic drugs, sulfa-containing antibiotics, progestogens, and synthetic estrogens. In these cases, the porphobilinogen (PBG) substrate for the deficient PBG deaminase is elevated in urine, and porphyrin concentrations in plasma and stool may be tested to exclude other porphyrin pathway disorders.
Porphyria cutanea tarda (PCT)
PCT is a chronic porphyria of the skin, associated with blistering cutaneous photosensitivity. Photosensitization results from accumulated, photo-excited porphyrins that are reactive. It is important to shield dark and light-pigmented skin from sunlight, as sunscreen does not protect from damaging wavelengths in patients with PCT. The familial form results from a genetic defect in uroporphyrinogen III decarboxylase. Patients with monoallelic or biallelic defects in the HFE gene (associated with the iron overload disorder hemochromatosis) may also show symptoms of PCT. Acquired PCT is associated with alcohol, smoking, hepatitis C, estrogens, and HIV. A patient may be initially asymptomatic but develop skin lesions when they take drugs that induce porphyrin synthesis or drink excessive alcohol. Porphyrin intermediates accumulate and react with oxygen to produce reactive oxygen species (ROS) that cause skin damage.
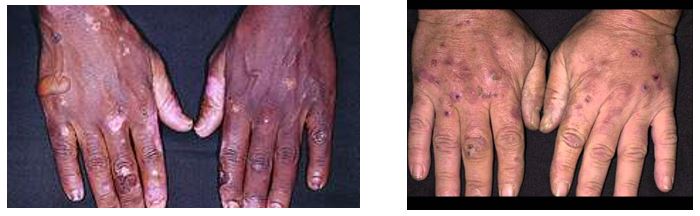
Figure 4.
Photodynamic diagnosis and therapy (PDT)
Properties of porphyrins have been exploited in photodynamic diagnosis (e.g. to reveal location of cancer cells) and photodynamic therapy (PDT), where porphyrin photosensitizers are activated to promote a selective cytotoxic effect on cancer cells. PDT is used commonly to treat pre-cancerous skin conditions. In “blue light therapy,” aminolevulinic acid (ALA) is first applied topically to affected areas. Abnormal skin cells (to a much greater extent than surrounding normal cells) convert ALA to protoporphyrin IX (Pp IX), the final intermediate in the porphyrin pathway to heme. When skin is subjected to blue fluorescent light of appropriate wavelength, the Pp IX absorbs energy and produces oxygen free radicals that damage and kill the abnormal skin cells.
SLO 3: Describe the importance of heme degradation in the development of hyperbilirubinemia.
Heme is catabolized first to biliverdin, a green pigment, which is converted to bilirubin, a yellowish red pigment that must be excreted to avoid toxic effects of hyperbilirubinemia. Bilirubin is processed in the liver for eventual excretion via the intestine or kidney. This section discusses localized heme catabolism in skin with bruises, basics of hepatic bilirubin conjugation to facilitate excretion, and lastly, consequences of elevated bilirubin levels in utero and in the newborn.
Bruises
When capillaries in the skin are damaged, blood extravasates, hemoglobin is released from damaged red blood cells, and white blood cells eventually clear the damage. In tissue macrophages, globin is degraded by proteolysis, and heme is catabolized to biliverdin by heme oxygenase, which opens the heme ring, releasing ferric iron (Fe3+) and carbon monoxide. Biliverdin is then converted to bilirubin by biliverdin reductase, and the iron is taken into ferritin, an iron storage protein that may become part of hemosiderin, contributing to a yellow-brown color in bruises. The presence of hemoglobin (red-blue) and the heme catabolites biliverdin (green) and bilirubin (yellow-red) typically follows the color-changing sequence: pink/red -> purple/blue/black-> violet/green/dark yellow -> pale yellow.
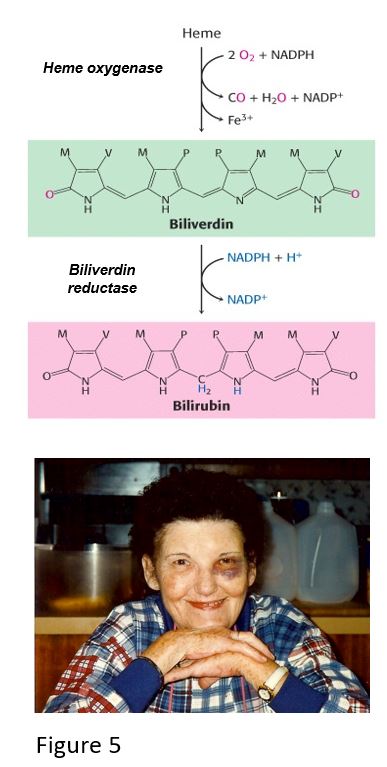
Sometimes blood extravasating from a damaged capillary can leak into tissue near the site of trauma, causing discoloration. Hemorrhagic “blotching” greater than 1 cm, from blood pooling under the skin or mucus membranes, is called ecchymosis. An example of this is periorbital ecchymosis, which may appear as if the person has a “black eye.”
With intravascular lysis of red blood cells, the hemoglobin (Hb) tetramer binds a haptoglobin (Hp) tetramer. The Hb/Hp complex protects the body against potentially harmful oxidative damage by free hemoglobin, heme and iron until globin is degraded and heme is converted to bilirubin, which is then processed in the liver for excretion. Haptoglobin is depleted when excess hemoglobin is released during red blood cell lysis in hemolytic anemia. Consequently, haptoglobin is one of the analytes assayed in screening and monitoring for intravascular hemolytic anemia. Haptoglobin levels may also increase during inflammation and infection, warranting its designation as an acute-phase reactant.
Hepatic bilirubin metabolism
Bilirubin is transported in the blood by serum albumin. Bilirubin enters the hepatocyte via a form of organic anion transporter polypeptide (OATP) and binds glutathione S-transferase (GST, formerly called ligandin), which concentrates the unconjugated, hydrophobic bilirubin in the cell. Conjugation of bilirubin with glucuronic acid via UDP-glucuronosyl transferase (UGT), results in the more hydrophilic bilirubin glucuronide and bilirubin diglucuronide that are refered to as conjugated bilirubin. Conjugation of bilirubin facilitates its transport into the biliary canaliculi via multiple drug resistance associated protein-2 (MRP2) pump. The related MRP3 on the sinusoidal membrane pumps conjugated bilirubin back into the blood in times of conjugated bilirubin excess, for example when there is a defect in MRP2 that prevents efficient excretion of conjugated bilirubin from the hepatocyte into the bile.
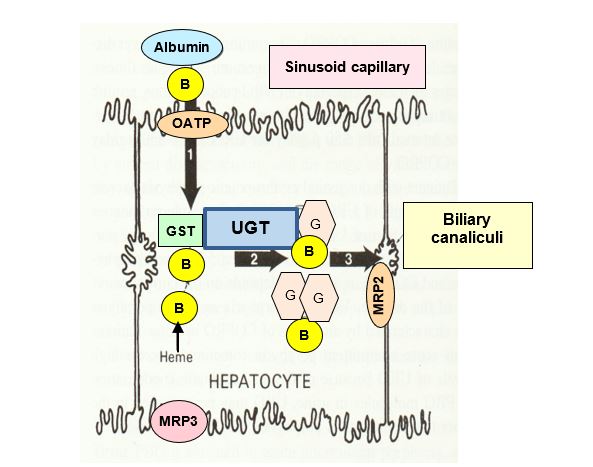
Figure 6.
Defects in the UDP-glucuronosyl transferase (UGT) gene can cause elevated bilirubin. A relatively common defect is associated with reduced UGT expression, frequently caused by a promoter mutation in the UGT1A1 gene. Patients with this defect have Gilbert syndrome and may be asymptomatic or present with occasional periods of mild hyperbilirubinemia. Children with the rare genetic disorder Crigler-Najjar syndrome, either do not have a detectable level of UGT (Type 1, very severe) or have very low levels of UGT (Type 2). Reduced transport of conjugated bilirubin into the bile is caused by a defect in the gene encoding MRP2 (Dubin-Johnson syndrome). Processing and excretion of bilirubin, relevance to disorders, and changes in analytes will be revisited in more detail in the liver section of another foundations course.
Note that UGT is not a cytochrome P450 enzyme, it has multiple isoforms, and is a Phase 2 enzyme involved in processing several endogenous metabolites and xenobiotics.
Neonatal jaundice
In utero, fetal bilirubin is processed by the maternal liver, so under normal circumstances, toxicity from bilirubin is avoided. However, after birth, processing of bilirubin by the newborn is dependent on the expression of UDP-glucuronosyl transferase (UGT), which is one of the last enzymes to be expressed in the developing fetal liver. Until UGT is produced adequately in a newborn (especially if premature), they will present with jaundice. Newborn infants with jaundice may be treated by exposure to bili-lights within a certain range of wavelengths, in order to isomerize bilirubin to a more hydrophilic form that is excreted more easily. If bili-lights are unavailable in a healthcare facility, limited exposure to sunlight may be recommended. In addition to the UGT deficiency described above, there are other causes of neonatal jaundice, such as dehydration and breastfeeding failure jaundice.

Figure 7.
Hemolytic disease of the fetus and newborn
The most common situation where fetal hemolytic anemia may arise is with Rh incompatibility (also called “Rh disease”). This disorder can occurs if the mother lacks the RhD red blood cell antigen (RhD negative) and the father and fetus are RhD positive. The Rh-negative mother is at risk of exposure to the fetal RhD antigen during her first pregnancy with an RhD-positive fetus, especially during the birthing process. The mother mounts an immunological response to fetal RhD antigen, and her subsequent RhD-positive pregnancies will be at risk for damage to the fetus.
During pregnancy, if maternal IgG antibodies against fetal red blood cell antigens cross the placenta, this can cause lysis of circulating fetal red blood cells, releasing large amounts of fetal hemoglobin. The excess bilirubin from heme degradation, may exceed the level that the mother can process and excrete, thereby exposing the fetus to a toxic level of bilirubin. In addition, as a consequence of a fetal red blood cell deficit, immature fetal red blood cells called erythroblasts may be present in the fetal circulation, a condition called erythroblastosis fetalis. Severe cases may result in fetal death in utero, however fetal hyperbilirubinemia may lead to a range of consequences in the future child, from mild hearing loss to severe intellectual disability. A rare but serious form of brain damage called kernicterus, results from the chronic and permanent consequences of bilirubin-induced neurologic dysfunction (BIND).
As a preventive measure, an anti-RhD antibody drug (Rhogam) is given to the mother during her first and subsequent pregnancies to inhibit the mother’s immune system from being stimulated with the RhD fetal antigen.
Note that the Rh protein complex is unrelated to the glycosphingolipid ABO blood group antigens.

Figure 8.
Feedback: