
6 Introduction to Diabetes Mellitus
Session Learning Objective 1: Discuss the differences between Type 1 Diabetes Mellitus (T1DM) and Type 2 Diabetes Mellitus (T2DM)
Session Learning Objective 2: Discuss the health risks associated with diabetic ketoacidosis in T1DM
Session Learning Objective 3: Discuss the molecular events associated with the development of T2DM
Session Learning Objective 4: Recall the associated health risks of DM
.
SLO1: Discuss the differences between Type 1 and Type 2 Diabetes Mellitus
Diabetes Mellitus is a very common, very complex disease that you will learn much more about during this block and subsequent blocks. A key feature of both diseases is impaired glucose homeostasis (a key component of carbohydrate metabolism) resulting in hyperglycemia (high serum blood sugar). For both diseases a key cause of hyperglycemia is inadequate insulin signaling through the insulin receptor, but the mechanisms are very different.
Type 1 Diabetes Mellitus (T1DM or DM1)
In patients with DM1, inadequate insulin signaling is caused by an absolute insulin deficiency. Plasma levels of insulin and c-peptide are undetectable or very low due to destruction of pancreatic β-cells. The most common cause of β-cell destruction is autoimmune, and many patients with DM1 will have high levels of autoantibodies such as GAD65, ICA, IAA or IA-2 (you do not need to know these exact tests, just know autoantibody testing is used to diagnose DM1).
The onset of DM1 is commonly very rapid and severe. Symptoms often include frequent urination (polyuria), thirst (polydipsia), weight loss despite an increased appetite (but sometimes nausea, vomiting and abdominal pain), and blurred vision. Many patients initially present in diabetic ketoacidosis (DKA, see below). DM1 is more commonly diagnosed in children or young adults but can occur at any age. The incidence of DM1 is increasing worldwide and the cause of this remains an important research area (eg. Covid-19 infection is now possibly implicated). DM1 is more common in people with first-degree relatives with DM1, but the genetically attributable increased risk is modest (incidence increases roughly from 30/100000 to 400/100,000 people). DM1 accounts for about 5% (~1.3 million) of all patients with diabetes mellitus in the US. Patients with DM1 need insulin therapy to maintain glucose homeostasis. A small fraction of patients with DM1 receive either pancreas transplantation or islet cell transplantation, but these therapies can normalize blood glucose regulation. Stem cell therapy remains an active area of research as a cure for DM1.
Type 2 Diabetes Mellitus (T2DM or DM2)
In type 2 diabetes mellitus (T2DM or DM2), inadequate insulin signaling is caused by insulin resistance and relative insulin deficiency. At the time of diagnosis plasma insulin levels and c-peptide levels are normal or even high, but because of insulin resistance those levels are insufficient and so serum glucose levels become too high. The most common risk factor for insulin resistance is intra-abdominal obesity which results in an excess of inflammatory signals (see below in SLO3 for cellular details). β-cell damage also contributes to the development of DM2 (and a gradual loss of β-cell function is thought to pre-date the diagnosis by as much as a decade). The mechanism is not usually autoimmune (autoantibody titers are low or negative). Mechanisms include amyloid deposition, inflammation and mitochondrial dysfunction among others, and this remains an active research area.
The onset of DM2 is commonly slow and symptoms (polyuria, polydipsia, weight loss, blurred vision) are milder and progress gradually. Many ‘asymptomatic’ patients are diagnosed following a routine screening blood test (Hemoglobin A1c >6.5%), and it is estimated that more than 10% of people with DM2 in the US are undiagnosed. DM2 is more commonly diagnosed in adults although the diagnosis in children and young adults is becoming more common. The incidence of DM2 continues to increase worldwide in parallel with increased prevalence of obesity. Patients with DM2 frequently have a positive family history and the disease has strong, complex, multi-gene heritability (non-mendelian). DM2 accounts for about 90% (~ 30 million) of all US patients with diabetes mellitus. Patients with DM2 should be treated with lifestyle measures to aid with weight loss, insulin sensitizers to decrease insulin resistance, insulin secretagogues to help β-cell insulin production increase, medications to help excrete glucose in the urine, as well as medications to help with weight loss. Insulin therapy may be needed as β-cell destruction progresses over years but is often used too early for patients with DM2 when better options exist.
Your math skills should tell you that leaves about 5% of people with diabetes mellitus that do not have classical DM1 or DM2, although the pathophysiology of hyperglycemia involves the same mechanisms. The list of these disorders is very long and includes hormonal disorders (causing excess cortisol, growth hormone or glucagon), pancreatic disorders causing β-cell damage (chronic pancreatitis, hemochromatosis, cystic fibrosis), and monogenic DM disorders (eg. glucokinase mutations).
Diabetes Mellitus and carbohydrate metabolism
Pause. From your previous readings this week you should be able to predict how carbohydrate metabolism is affected by diabetes mellitus
Decreased Insulin Signaling:
- Glucose uptake (also called disposal). Digestion and absorption of carbohydrate is unaffected (not always true you will learn later about diabetic gastroparesis), but without insulin signaling glucose transport via GLUT4 into adipocytes and myocytes is reduced and blood glucose following a meal is too high. This is called impaired glucose tolerance (IGT). The high glucose after meals (post-prandial) results in increased glucose transport through other GLUT (eg. not insulin-dependent GLUT4) into cells that can be damaged by higher cytoplasmic glucose levels (more detail below).
- Glycogen storage and gluconeogenesis. Impaired insulin signaling results in decreased glycogen synthesis and increased glycogen breakdown (you should know this mechanism now related to increased phosphorylation (because kinase activity higher than phosphatase activity) of glycogen synthase and glycogen phosphorylase. In the liver this effect on glycogen storage contributes to hyperglycemia because glucose is released into the circulation in excess of what the body requires. The same is true for gluconeogenesis. Insufficient insulin signaling favors increased gluconeogenesis causing the liver (and kidneys) to make glucose from lactate, amino acids and glycerol (you should be able to describe this mechanism related to decreased levels of cytoplasmic fructose 2,6, bisphosphate). When these processes happen overnight the result is impaired fasting glucose (IFG).
- Glucagon. Glucagon is produced by α-cells in the pancreatic islet. These cells are inhibited by local secretion of insulin by β-cells. When insulin signaling is inadequate glucagon levels increase and high glucagon levels are common in patients with diabetes mellitus. High glucagon will worsen the effect of low insulin signaling on glycogen breakdown and gluconeogenesis.
- Other hormones. Epinephrine can increase for a variety of reasons (eg. dehydration) which will also worsen hyperglycemia (though more profound effects are on adipocytes as you will hear later). Cortisol can increase for a variety of reasons (eg. dehydration, infection, nausea) which will worsen hyperglycemia. The adipocyte hormone leptin decreases (from increased lipolysis) in DM1 which causes increased appetite, decreased energy expenditure and changes in non-insulin mediated glucose uptake which all favor hyperglycemia. The adipocyte hormone adiponectin decreases in the setting of obesity and DM2 and contribute to decreased insulin sensitivity
SLO2: Discuss the health risks associated with diabetic ketoacidosis in DM1
While this SLO focuses on DKA, you should understand the following two hyperglycemic emergencies.
- Diabetic Ketoacidosis (DKA). This emergency occurs most commonly in patients with DM1. Risk factors are inadequate insulin treatment (eg. cost, adherence, equipment failure, failed absorption, etc.) and acute stressors (eg. infections, trauma, etc.) and DKA often occurs in a time period of hours. Blood glucose levels are usually very high (400-600 mg/dL) due to decreased glucose uptake and increased glycogen breakdown and gluconeogenesis (see above). Dehydration from polyuria causes increased glucose reabsorption and worsens the hyperglycemia. Very high plasma glucose becomes osmotically active in plasma which shifts water out of the cellular cytoplasm and dilutes serum sodium (hyponatremia with normal plasma osmolality, which minimizes low sodium symptoms). Electrolyte abnormalities such as low potassium (hypokalemia) are common. Dehydration and acidosis can cause changes in blood vessels in the brain that cause cerebral edema, especially when rehydration and blood sugar lowering with insulin is rapid. The ketosis comes from uninhibited triglyceride lipolysis in adipose tissue which is converted to ketones by the liver (you will learn about this next week). Emergency treatment focuses on fluid and electrolyte repletion, blood glucose lowering with insulin and correction of acidosis. If the level is low, potassium has to be repleted before insulin is given because the sudden shift of glucose into the cells also shifts potassium into cells (not via GLUT but via a Na+/H+ anti-porter and then the Na+/K+ ATP’ase pump) which can cause life-threatening hypokalemia (causes arrhythmia).
- Hyperglycemic Hyperosmolar State (HHS). This emergency occurs most commonly in patients with DM2. Risk factors are stressors (infection, myocardial infarction, stroke, injury, pain, medications) and non-adherence with medications. Blood glucose levels can be extremely high (600-1200 mg/dL) for the same reasons as in DKA, except that HHS usually occurs over a period of days of progressive illness. Patients with HHS are hyperosmolar and the serum sodium may be high (hypernatremia) or artificially normal due to the hyperglycemia (see above). Patients may also be acidotic and this is often a mixed disorder with lactic acidosis and (milder) ketoacidosis. Insulin signaling is not usually low enough to cause unrestrained lipolysis resulting in DKA (more next week), but after days of severe hyperglycemia β-cells can develop glucotoxicity which acutely shuts down insulin production and ketoacidosis can occur. Emergency treatment focuses on fluid and electrolyte repletion, blood glucose lowering with insulin and identification and management of underlying conditions causing the HHS (eg. infection). Morbidity rates and mortality rates for patients with HHS remain high.
SLO3: Discuss the molecular events associated with the development of DM2
Identification of the links between abdominal obesity and insulin resistance and β-cell damage remains a major goal of obesity/diabetes research. Evidence supports inflammation caused by excessive or ectopic deposition of triglyceride, as well as increased signaling from free fatty acids as playing major roles.
DM2 as a chronic inflammatory disease. Increased abdominal (also called central or visceral) adipose tissue deposition eventually results in macrophage infiltration within adipocytes which then release systemic cytokines such as tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6). This low-grade systemic inflammation has several effects.
- Systemic inflammation activates intracellular inflammatory pathways, such as nuclear factor kappa B (NF-kB). Once activated, NF-kB induces the expression of molecules involved in inflammation and oxidative stress, including JNK which inhibits insulin signaling.
- Intracellular molecules such as JNK cause increased serine phosphorylation which prevents insulin-mediated tyrosine phosphorylation resulting in insulin resistance. Serine phosphorylation can occur on various components of the insulin signaling cascade, including insulin receptor substrate (IRS) proteins. When IRS proteins are serine phosphorylated this inhibits their ability to activate PI3K, and insulin signaling decreases.
- Chronically increased serum free fatty acids also stimulate NF-kB, as well as causing endoplasmic reticulum (ER) stress. ER stress can lead to the activation of the unfolded protein response (UPR), which is a cellular defense mechanism that aims to restore ER homeostasis. However, chronic ER stress can lead to the activation of pro-inflammatory pathways contributing to insulin resistance.
- Altered fatty acid metabolism also increase the production of modified phospholipids (ceramides) that activate specific protein phosphatases. These phosphatases dephosphorylate active tyrosine-phosphorylated IRS proteins and decrease insulin signaling.
- Adipose tissue inflammation decreases the secretion of the adipocyte hormone adiponectin. This hormone increases glucose uptake and metabolism through the AMP-activated kinase pathways and normally counteracts insulin resistance. Low adiponectin can contribute to hyperglycemia.
Ectopic lipid deposition. In the setting of obesity, triglycerides and free (non-esterified) fatty acids begin to accumulate in other tissues that do not normally store triglycerides. These include skeletal muscle, gonads (contributes to polycystic ovarian disease), pancreas (contributing to β-cell damage), heart (contributing to cardiac dysfunction) and liver (causing hepatic steatosis and steatohepatitis). Triglyceride deposition in these tissues contributes to organ dysfunction and also causes local and systemic inflammation which can worsen insulin resistance.
Genetics and insulin resistance. The risk of developing DM2 can be inherited. The genetics are complex (eg. non-mendelian involving multiple genes that individually convey only low risk) and intricately involved with nutrition, activity and social determinants of health. An enormous amount of research has tried to identify candidate susceptibility genes, and while several genes have been shown to have small effects, there is currently no single defect that explains insulin resistance (eg. if you study 100 patients with the same level of body adiposity and 50 are insulin resistant and 50 are insulin sensitive, there is no single gene mutation that explains the 50 patients with insulin resistance).
SLO4: Recall the associated health risks of DM
Diabetes mellitus is associated with very significant long-term complications that result in most of the morbidity and mortality from the disease. These include both microvascular and macrovascular complications. The classical microvascular complications are retinopathy (edema and vascular changes in the retina leading to visual impairment), nephropathy (damage to the glomerulus which results in proteinuria and chronic kidney failure), and peripheral neuropathy (results in dysesthesias and eventually sensory loss which predisposes to injury, poor wound healing and lower extremity amputation). Macrovascular complications result from an increased risk of atherosclerosis of vessels of all sizes along the arterial tree. This increases the risk of myocardial infarction (heart attack), cerebrovascular disease (stroke) and peripheral vascular disease. Many studies have shown that improved glycemic control reduces the risk of developing both microvascular and macrovascular disease
Biochemistry of microvascular disease
While this topic remains an active research area there are multiple mechanisms known to link hyperglycemia to microvascular disease.
- Advanced glycation end products (AGEs) are formed by the non-enzymatic reaction between reducing sugars (eg. glucose) and amino groups of proteins, lipids, and nucleic acids. In DM, hyperglycemia leads to the accumulation of AGEs, which can modify proteins and alter their function. The accumulation of AGEs in the basement membrane of blood vessels can lead to thickening and reduced tissue perfusion.
- Cytoplasmic hyperglycemia increases oxidative stress from increased flux of electrons through the mitochondrial electron transport chain resulting in enhanced production of superoxide. Oxidative stress can damage cellular components such as proteins, lipids, and DNA. The resulting oxidative damage can lead to endothelial dysfunction, increased vascular permeability, and inflammation.
- Hyperglycemia can activate protein kinase C (PKC), which is involved in various cellular processes, including angiogenesis and vascular permeability. PKC activation leads to increased production of cytokines and growth factors, promoting inflammation and inappropriate angiogenesis.
- Hyperglycemia also leads to intracellular sorbitol accumulation. Sorbitol is normally metabolized to fructose by the enzyme sorbitol dehydrogenase (SDH), but chronic hyperglycemia overwhelms this pathway. Sorbitol accumulation can lead to osmotic stress and cellular damage, particularly in tissues with limited or no SDH, such as the retina, nerves, and kidneys. The accumulation of sorbitol can lead to alterations in cellular function, including impaired mitochondrial function, increased oxidative stress, and reduced energy production.
Hemoglobin A1c is an example of an AGE. Glucose reacts with a valine on the hemoglobin β-chain. Because the lifetime of a red cell is about 90 days, the level of hemoglobin A1c provides an indication of average blood glucose levels over the previous 3 months and is used to diagnose diabetes mellitus and monitor glycemic control in patients with DM1 or DM2.
Biochemistry of macrovascular disease
Atherosclerosis and macrovascular disease is a giant topic and we will only touch on a few mechanisms here related to hyperglycemia. Those mechanisms are largely the same, relying on AGEs, oxidative stress and inflammation except the target tissues now are endothelial cells (the inner lining of blood vessels) and smooth muscle cells (a component of the arterial wall). We will discuss only two specific mechanisms beyond what was described above.
- In the vasculature, inflammation induced oxidative stress alters low-density lipoproteins (LDL, wait until next week) to form oxidized LDL, which can stimulate the expression of adhesion molecules on endothelial cells and promotes infiltration of monocytes (white blood cells already primed by the systemic inflammation, called macrophages once they leave the vasculature), leading to the formation of foam cells (dying macrophages) and the development of atherosclerotic plaques.
- Endothelial dysfunction decreases nitric oxide (NO) production and bioavailability. NO is a potent vasodilator and inhibitor of platelet aggregation, and reduced production leads to vasoconstriction, increased platelet aggregation, and thrombus formation, promoting the development of atherosclerosis.
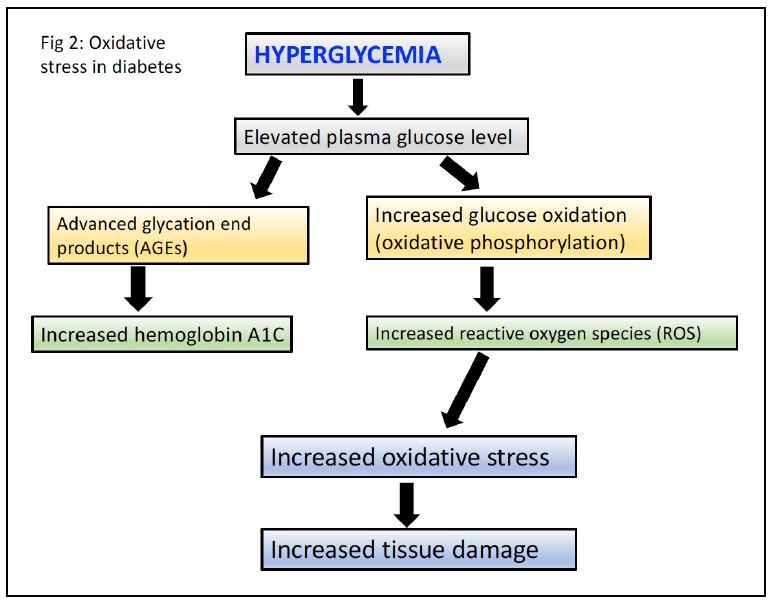
Figure 1.
Practice questions:
1. Can you draw the reaction catalyzed by sorbitol dehydrogenase? Why is this enzyme important?
3. Explain the interaction between high LDL levels and hyperglycemia in DM2?
4. Explain the differences between insulin levels and insulin signaling in DM1 and DM2?
5. Hypothesize how insulin sensitizers might be used in the treatment of DM2?
Feedback