
11 Nitrogen Metabolism, Urea Cycle
The goal of this chapter is to help students understand the concept of nitrogen balance, and how the body gets rid of excess ammonia (NH3) or ammonium ions (NH4+) when amino acids are catabolized.
Session Learning Objectives:
SLO4: Discuss the diagnostic significance of aspartate aminotransferase and alanine aminotransferase (AST, ALT). [Covered in Class].
Overview: Nitrogen is consumed mostly as protein in the human diet. The successive actions of proteolytic enzymes in the stomach and small intestine hydrolyze proteins to smaller oligopeptides and amino acids. The intestinal brush border cells contain oligopeptidase enzymes which complete the digestion process. Proteins must be degraded to amino acids or very short peptides before they can be absorbed. After absorption from the small intestine into the blood stream, amino acids have three metabolic fates in the body:
1. Synthesis of tissue proteins
2. Synthesis of other important constituents of the body
3. Catabolism and excretion of the products
SLO1: Define the concept of nitrogen balance and explain the role of protein degradation in normal nutrition and disease states.
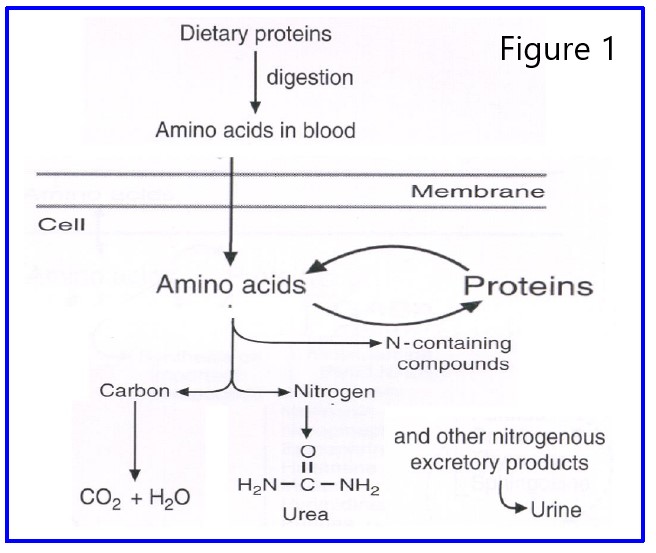
Metabolic Fate of Nitrogen: Figure 1 illustrates the metabolic fate of nitrogen ingested by a mammal. Ingested amino acids enter the body’s amino acid pool. This pool is rather small because incoming amino acids are rapidly shuffled off to other places. Their principal fate is incorporation into proteins. Amino acids may also contribute their carbon skeletons to synthesis of various TCA cycle intermediates or acetyl CoA, and their nitrogen can contribute either to the synthesis of other nitrogenous compounds (purines, pyrimidines, other amino acids, porphyrins, etc.) or to urea, which is excreted. The excretion of urea is the principal way the human body gets rid of excess nitrogen. A small amount of nitrogen is also lost by excretion of other nitrogenous compounds in the feces, through the skin, and during respiration.
Ammonia has an important place in the flow of nitrogen through the body. Ammonia from catabolites or other sources can be picked up for synthesis of nitrogen-containing molecules, or can combine with CO2 to form urea and be excreted.
NITROGEN BALANCE
Nitrogenous compounds, such as amino acids, continually enter and leave the human body. The normal, healthy adult maintains nitrogen equilibrium, assimilating about 5 grams of nitrogen per day and excreting the same amount. During infancy and childhood (growth) and during convalescence from certain diseases, the intake of nitrogen exceeds the amount excreted — this is referred to as positive nitrogen balance. During starvation or certain disease states, the output of nitrogen may exceed the amount taken in — a negative nitrogen balance. This represents a net loss of tissue protein due to catabolism of the constituent amino acids.
DEGRADATION OF AMINO ACIDS
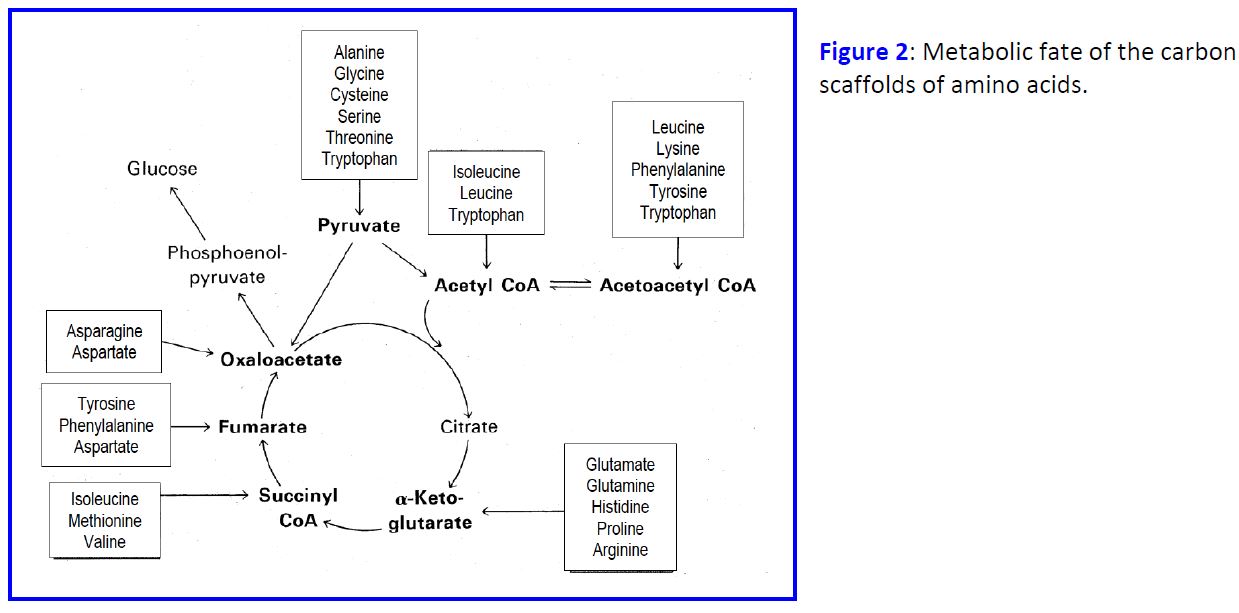
There is no storage polymer for excess amino acids like there is for excess glucose or fatty acids. Amino acids in excess of those required for synthesis of protein and other compounds are converted to major metabolic intermediates. The alpha-amino group is removed and the resulting carbon skeleton converted either to TCA cycle intermediates (glucogenic amino acids) or to acetyl-CoA (ketogenic amino acids). All amino acids can contribute to energy production through the TCA cycle or to fatty acid biosynthesis via citrate. Glucogenic amino acids are so named, because they can be used for gluconeogenesis in the liver; the ketogenic amino acids, on the other hand, are used to make ketones under these same conditions. Figure 2 summarizes the fate of the carbon skeletons.
SLO2: Describe the metabolism of nitrogen using aminotransferases, glutamate dehydrogenase, glutamine synthetase and glutaminase.
Transamination Reaction
The major mechanism for removing alpha-amino groups from amino acids is via transamination (Figure 3). Transaminases (aminotransferases) catalyze the transfer of the alpha-amino group of an amino acid to the carbon of an alpha-keto acid. The most common alpha-keto acid used as an acceptor of amino groups in these reactions is alpha-ketoglutarate, forming glutamate. There are many different aminotransferases, specific for different amino acids, but almost all of these use alpha-ketoglutarate and L-glutamate as one of the substrate pairs.

Reactions catalyzed by glutamate dehydrogenase. This reaction (Figure 4) is fully reversible according to the needs of the cell, although in vivo it probably functions primarily to deaminate glutamate, especially considering that many humans (Americans at least) consume an excess of protein and burn it for energy. Note that ammonia is produced in this oxidation reaction. The reaction, as written, takes place in the mitochondrial matrix and uses NAD+ as the preferred cofactor.
In the cytosol, the enzyme uses small amounts of ammonia present and NADPH as the cofactor for glutamate synthesis (the reverse of the reaction shown). Working together with transaminases, this can yield nonessential amino acids.

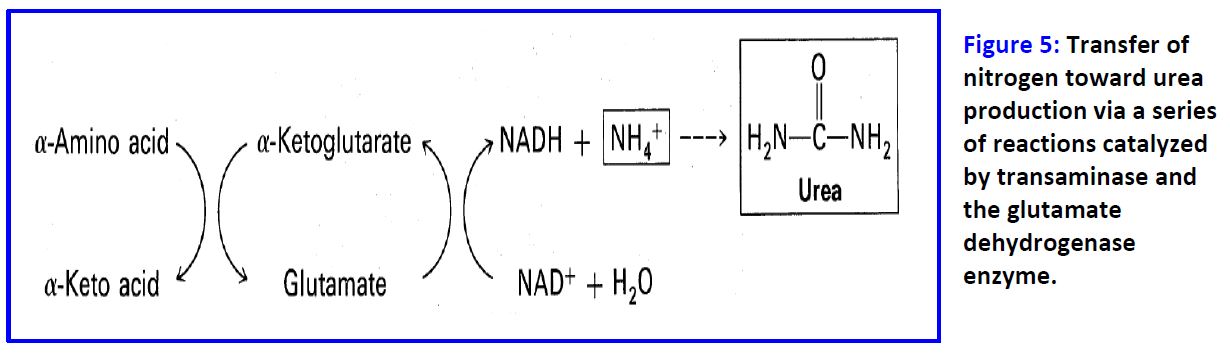
Transaminases funnel alpha-amino groups from most amino acids into glutamate. Glutamate is then transported into the mitochondrion and oxidatively deaminated to produce an ammonium ion, which is converted into urea and excreted (Figure 5). Transaminases also function in synthesis of nonessential amino acids, using the amino group of glutamate that is synthesized from alpha-ketoglutarate and ammonia.
Source of ammonia NH3/NH4+:
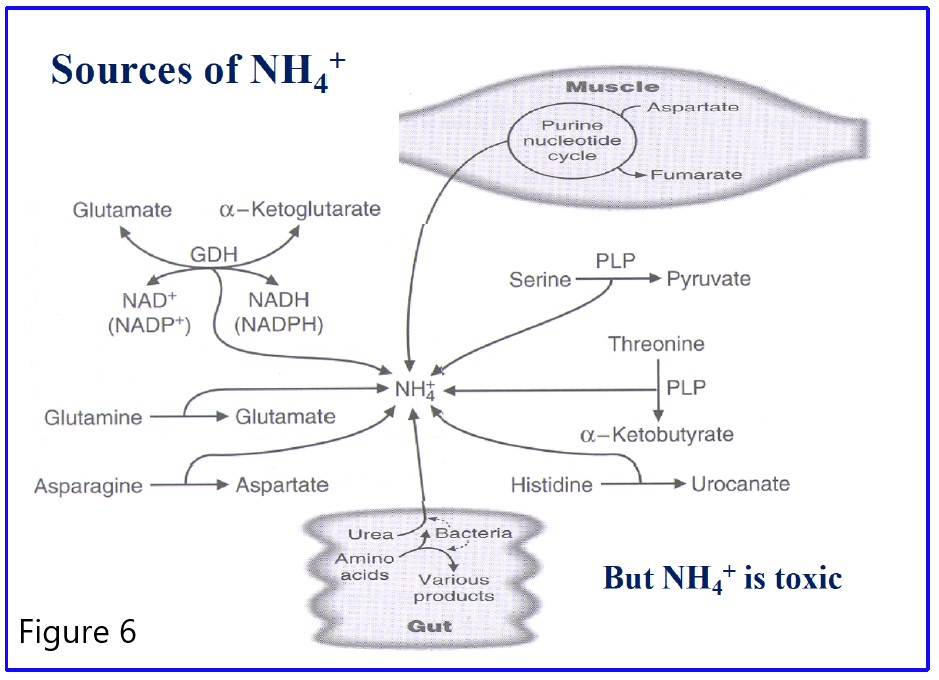
Figure 6: Summary of the sources of NH4+ for the urea cycle. All reactions are irreversible except glutamate dehydrogenase (GDH). Only the dehydratase reactions which produce NH4+ from serine and threonine require pyridoxal phosphate (PLP) as a cofactor. In the liver, NH4+ generated is converted to urea. Note the contribution of the gut bacteria.
SLO3: Describe the significance of the urea cycle in removing nitrogen and the presentation of hyperammonemia with defects in urea cycle enzymes.
NITROGEN ELIMINATION AND THE UREA CYCLE
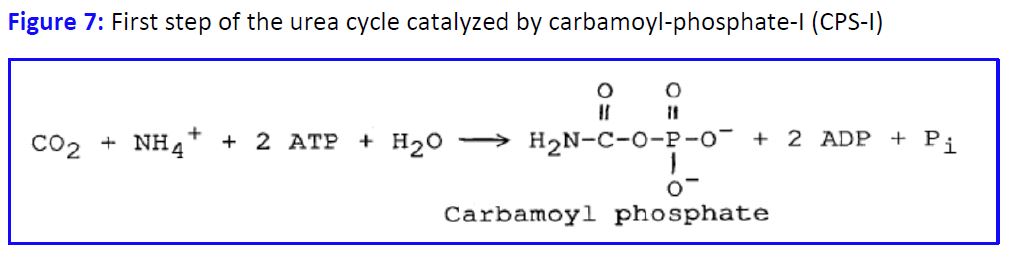
Nitrogen is excreted from humans mainly as urea, uric acid and ammonia. Ammonia is toxic at high concentrations (see below), and uric acid is poorly soluble, so nitrogen is mostly eliminated as urea. The urea cycle begins with the incorporation of ammonia into carbamoyl phosphate (Fig. 7).
The Urea Cycle (Fig 8): Part of the reaction occurs in the mitochondria, the rest in the cytoplasm. The cycle proceeds through a series of intermediates, resulting in urea being eliminated from arginine.
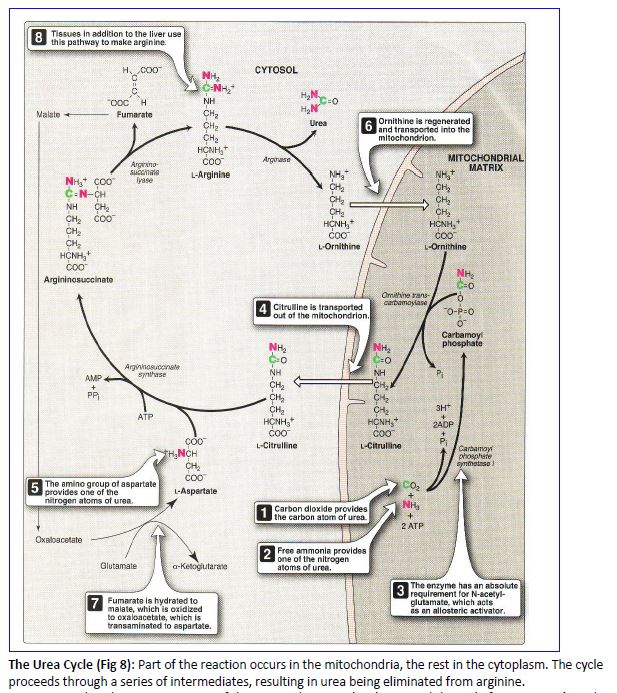
Note that the nitrogen atoms of the excreted urea molecule entered the cycle from ammonia and aspartic acid. Thus, in considering the flow of nitrogen from an amino acid catabolized to urea, we have the following scheme in Fig 9.

The reactions of the urea cycle are distributed between the cytosol and the mitochondrial matrix. This sequesters ammonia produced by the glutamate dehydrogenase reaction inside the mitochondrion, where it can immediately feed into the urea cycle. Note that transport systems are present in the inner mitochondrial membrane for moving intermediates between the cytosol and the mitochondrial matrix.

REGULATION OF THE UREA CYCLE
Long term adaptive response: A high protein diet leads to elevated serum glucagon, which in turn stimulates transcription of the genes encoding the urea cycle enzymes (Figure 11).

Short term regulation: Carbamoyl phosphate synthetase I requires N-acetylglutamic acid as an allosteric activator. The activity of the enzyme that catalyzes the synthesis of N-acetylglutamic acid from acetyl CoA and glutamic acid in the liver is stimulated in response to the arginine contained in a high protein meal. N-acetylglutamate signals dietary protein status, controlling the urea cycle through regulation of carbamoyl phosphate synthesis (Figure 10).
High protein diets lead to elevated glucagon Levels that promotes transcription of the genes encoding the Urea Cycle enzymes.
In the scheme above (Figure 11), alanine is converted to glucose and urea via the following reactions:
1. Alanine – key gluconeogenic amino acid is transaminated to form pyruvate which is converted to glucose.
2. The nitrogen now in glutamate can be released as NH4+ (2) or transferred to oxaloacetate to form aspartate (3)
NH4+ and aspartate enter the urea cycle which produces urea.
CONGENITAL HYPERAMMONEMIA
This is a group of diseases characterized by elevated ammonia levels, often accompanied by episodic vomiting, hepatomegaly, intolerance to dietary protein, lethargy, retardation and in severe cases coma and death shortly after birth. Deficiency in any of the enzymes in the urea cvcle can produce these symptoms. Several of the best known examples are discussed below.
Carbamvl Phosphate Svnthetase Deficiency is an autosomal recessive disease with severe hyperammonemia and a rapidly progressive fatal course shortly after birth. Such a complete absence of any urea cycle enzyme is fatal, since there is no alternate pathway for urea synthesis.
Ornithine Transcarbamylase Deficiencv (OTC) is an x-linked disorder with a course similar to the above in males with complete absence of the enzyme; those with reduced activity can be managed with a low protein diet. Heterozygous females can have variable symptoms.
Citrullinemia, argininosuccinic aciduria, and argininemia are all also known; in each case, there is blockage of the step following the accumulated urea cycle intermediate, combined with hyperammonemia.
Ammonia toxicity is speculated to be a common factor to all of these disorders, and is caused by reduced levels of alpha-ketoglutarate inside the mitochondria, due to a shift in the equilibrium of the glutamate dehydrogenase reaction. This results in reduced TCA cycle activity and lowered ATP synthesis. The effects of reduced oxidative metabolism are particularly deleterious to brain function. These diseases have a precipitous course immediately after birth, because maternal metabolism no longer detoxifies the blood of the fetus.
Practice questions:
1. What is the rate limiting step of urea biosynthesis?
2. How is nitrogen originating from the NH2 of amino acids targeted for degradation moved around to be made available for urea synthesis
3. Outline the mechanisms by which urea synthesis is regulated. Why does it make sense for glucagon to activate gluconeogenesis and the urea cycle simultaneously?
4. What are the health consequences of deficiencies in the enzymes of the urea cycle?
Feedback: