
11 Neuroendocrinology of Energy Homeostasis
Consumed food is stored as energy in the form of two primary macromolecules, glycogen and triglycerides, both of which are created through anabolic processes. Glycogen reserves, stored in the liver and skeletal muscles, have a finite capacity. In contrast, triglyceride reserves in adipose tissue have an almost unlimited capacity.
Between periods of energy consumption, glycogen and triglycerides are catabolized to release a continuous supply of glucose for use by the brain, as well as glucose, ketones, and fatty acids for the rest of the body. For energy reserves to remain in balance, energy consumption must ultimately equal energy expenditure. When energy consumption exceeds expenditure, adiposity will increase. If energy consumption fails to meet energy demands, starvation will result.
Multiple mechanisms collectively create a balance between energy consumption and energy expenditure (in both the short term and the long term). Some signaling pathways influence the digestion, absorption, and storage of consumed energy sources; other signaling pathways influence behaviors driving consumption; and some signaling pathways modulate internal metabolic processes that utilize energy. There is substantial overlap in signaling pathways involved in various components of energy homeostasis.
Here, we will examine the signaling pathways that regulate energy consumption and begin to examine thermoregulatory pathways involved in energy expenditure through adaptive thermogenesis. There is substantial overlap in the neuroendocrine signaling pathways involved in energy consumption and energy expenditure, all of which contribute to the maintenance of energy homeostasis.
Regulation of digestion, absorption, and storage of consumed energy is largely achieved through gastric hormone signaling pathways. Different types of food entering the gastrointestinal tract stimulate secretions of different hormones, which signal the presence of those different types of food and generate appropriate responses. Some of these gastric hormones modulate digestive processes by regulating the excretion of enzymes into the digestive tract, or by changing the rate at which ingested food is transported through the digestive tract. These gastric hormones released during digestive processes, as well as the nutrients absorbed during digestion, often operate at the level of the brain as well, where they regulate behaviors involved in energy consumption.
Certain regions of the brain, particularly the hypothalamus, respond to peripheral signals of energy availability by modulating food consumption and energy expenditure, either to replenish depleted energy stores (by promoting food consumption and reducing energy expenditure) or to prevent the accumulation of excess energy stores (by reducing food consumption and increasing energy expenditure). Orexigenic signals, such as ghrelin from the stomach, are released in response to low energy availability, such as during periods of fasting. Orexigenic signals promote appetite and drive feeding behaviors to replenish depleted energy stores while also reducing energy expenditure. These signals are often suppressed by anorexigenic signals, which promote satiety through appetite-reducing pathways and enhance energy expenditure to release excess energy. Short-term anorexigenic hormones are released in anticipation of impending food consumption and during digestion of consumed food, signaling the impending absorption of energy sources. These short-term anorexigenic hormones are often involved in aspects of digestion, absorption, and storage of consumed nutrients. In other words, the release of short-term anorexigenic hormones are closely linked to the feeding-fasting cycle; short-term anorexigenic signals increase throughout periods of food consumption, contribute to the termination of food consumption, and then decrease following food consumption, digestion, and storage as the stimulus for their release is terminated. In contrast, long-term anorexigenic hormones, such as leptin from adipose tissue, are signals released from energy reserves in proportion to the amount of energy stored in those reserves, thereby providing a metric of energy availability within the body. Thus, when food is consumed, short-term anorexigenic signaling increases, leading to termination of food consumption. As the newly consumed energy is expended over a period of fasting, there is a decrease in short-term anorexigenic signaling accompanied by an increase in orexigenic signaling. Throughout the feeding-fasting cycle, long-term anorexigenic signals are continually released. However, during periods of fasting, increasing orexigenic signaling will eventually overcome signaling by long-term anorexigenic hormones and promote the initiation of another meal.
Start WG07
The hypothalamus is the principal brain region responsible for integrating peripheral orexigenic and anorexigenic signals and conveying those signals to other brain regions involved in motivated food-seeking and feeding behaviors and the regulation of energy expenditure. Multiple, highly-interconnected regions and nuclei within the hypothalamus are involved in this process of signal integration, including medial hypothalamic nuclei such as the arcuate nucleus (ARC), paraventricular nucleus (PVN), ventromedial hypothalamic nucleus (VMH), and dorsomedial hypothalamic nucleus (DMH), as well as the lateral hypothalamus (LH).
While integration of peripheral orexigenic and anorexigenic signals by the PVN, VMH, DMH, and LH play vital roles in the maintenance of energy homeostasis, the ARC is the most prominent bridge through which peripheral signals influence the central regulation of energy homeostasis. After amalgamating peripheral orexigenic and anorexigenic signals, neurons in the ARC relay the integrated information to second-order neurons in the PVN, VMH, DMH, and LH, which project to extrahypothalamic circuits.
End WG07
Within the ARC, two types of neurons are particularly implicated in integrating peripheral signals of energy availability and transmitting those signals to other hypothalamic (and extrahypothalamic) regions. Firstly, the ARC contains anorexigenic POMC neurons, named for their expression of pro-opio-melanocortin (POMC). POMC is cleaved into numerous melanocortins, which serve as anorexigenic signal molecules. One of the most important anorexigenic signal molecules derived from POMC and used as an anorexigenic neurotransmitter by POMC neurons is α-melanocyte stimulating hormone (αMSH). αMSH activates melanocortin receptor types 3 and 4 (MC3R and MC4R). In addition to anorexigenic melanocortins, POMC neurons also express the anorexigenic signal molecule cocaine- and amphetamine-regulated transcript
(CART). Secondly, the ARC contains orexigenic NPY/AgRP neurons, named for their expression of neuropeptide Y (NPY) and agouti-related peptide (AgRP). NPY receptor types 1 and 5 (Y1R and Y5R) are most implicated in NPY-mediated stimulation of food consumption. In addition to NPY and AgRP, NPY/AgRP neurons also release GABA. POMC-derived anorexigenic signals are released tonically, with their release being modulated by orexigenic and anorexigenic signals. In contrast, signaling by NPY/ AgRP neurons is induced by orexigenic signals.
End WG08
Arcuate POMC and NPY/AgRP neurons project dendrites into the median eminence. The median eminence is a circumventricular organ with fenestrated capillaries that result in a ‘leaky’ blood brain barrier. This enables POMC neurons and NPY/AgRP neurons to efficiently sense the presence of peripheral hormones and nutrients in systemic circulation, even those that cannot permeate the normal blood-brain barrier. POMC neurons and NPY/AgRP neurons primarily project short axons and form local, intrahypothalamic connections. However, connections to some extrahypothalamic regions are also important in the regulation of energy homeostasis.
End WG09
The anorexigenic POMC neurons and orexigenic NPY/AgRP neurons are functionally antagonistic; POMC neuron signaling and NPY/AgRP neuron signaling yield almost exactly opposite effects on target neurons. In fact, AgRP is actually an antagonist at MC3R and MC4R, which enables orexigenic AgRP signaling by NPY/AgRP neurons to block anorexigenic αMSH signaling by POMC neurons. Not only do NPY/AgRP neurons block the effects of αMSH-mediated signaling by POMC neurons, but NPY/AgRP neurons actually directly inhibit POMC neurons with GABA.
The most prominent POMC neuron projections excite the PVN. While MC4R is expressed throughout the brain, the highest concentrations of MC4R are found within the PVN. Neurons in the PVN secrete regulatory neuropeptides (e.g., trophic hormone- releasing hormones) at the median eminence, and also regulate sympathetic efferents; the PVN is one central regulator of the sympathetic nervous system, and plays key roles in the generation of a sympathetic response. MC4R activation on PVN neurons promotes satiety and increases energy expenditure through increased metabolism.
These effects are mediated by the release of thyrotropin-releasing hormone (TRH) and corticotropin-releasing hormone (CRH) at the median eminence, activating the HPT and HPA axes, respectively, and by the release of brain-derived neurotropic factor (BDNF), among other signal molecules. MC4R activation on PVN neurons also increases signaling by sympathetic efferents; the PVN activates preganglionic sympathetic neurons of the intermediolateral nucleus in the upper thoracic spinal cord, increasing sympathetic tone, which further increases energy expenditure. The effects of the PVN on satiety and energy expenditure are crucial to normal regulation of energy homeostasis; destruction of the PVN results in hyperphagia and obesity. NPY/AgRP neurons inhibit PVN neurons, decreasing secretion of neuropeptides at the median eminence, and decreasing sympathetic tone. In other words, NPY/AgRP neurons antagonize the satiety-promoting and energy-expenditure promoting effects exerted by POMC neurons on the PVN. In fact, NPY/AgRP neuron signaling to the PVN alone is able to induce food consumption. This antagonism results in increased insulin secretion, decreased breakdown of triglycerides in adipose tissue, and decreased body temperature. In addition to PVN projections involved in the generation of a sympathetic response, PVN neurons also innervate arcuate neurons, enabling negative feedback regulation.
End WG10
POMC neurons and NPY/AgRP neurons also innervate the VMH, activity of which promotes satiety and energy expenditure. Similar to the PVN, POMC neuron signaling excites VMH neurons while NPY/AgRP neuron signaling inhibits VMH neurons. The importance of the VMH in promoting satiety and energy expenditure is evident in lesion studies, which demonstrate that destruction of the VMH, like destruction of the PVN, results in hyperphagia and obesity. Lesions to the VMH induce increased neural excitation of insulin secretion through autonomic innervation of the pancreas, and a general disruption of the balance between the sympathetic nervous system and parasympathetic nervous system. In animals with VMH lesions, the liver and adipose tissue fail to release energy stores during periods of fasting. Without the release of energy stores, animals are forced to eat more frequently to maintain sufficient levels of energy available in the bloodstream. Furthermore, such VMH-lesioned animals are unusually responsive to the positive (hedonic) aspects of food, and may even seek and consume foods that would otherwise be perceived as nauseating. Overall, VMH- lesioned animals rapidly become obese. However, if the connections to the pancreas are also lesioned, the increase in insulin is resolved, and obesity does not develop. Like the PVN, VMH neurons provide negative feedback to the arcuate nucleus. Along with POMC and NPY/AgRP neurons, the VMH also provides strong input to the LH.
End WG11
Neurons in the LH release multiple orexigenic signals. Some LH neurons produce the orexigenic transmitter orexin (also called hypocretin), which is also involved in the regulation of sleep by promoting wakefulness. There are two forms of orexin, orexin-A and orexin-B, and two types of orexin receptors, OX1R and OX2R, both of which are G- protein coupled. Orexin-A has equal affinity for both receptors, but orexin-B primarily binds OX2R, and with about five-times weaker affinity than orexin-A. Orexin is an important link between aspects of energy balance (both energy consumption and expenditure) and sleep regulation. Other LH neurons release the orexigenic signal melanin-concentrating hormone (MCH). However, while orexin promotes wakefulness, MCH promotes sleep, and is actually more active during sleep than wakefulness. Orexin and MCH neurons in the LH are very important appetite-promoters; their destruction results in hypophagia and a lean phenotype. LH neurons form local connections with medial hypothalamic nuclei, such as the PVN, at which orexin has an inhibitory influence, preventing PVN-mediated anorexigenic effects and thereby promoting appetite. LH neurons also project long axons to innervate extrahypothalamic regions throughout the brain that contribute to the generation of motivated food consumption, such as the ventral tegmental area (VTA), cerebral cortex, locus coeruleus, thalamus, reticular formation, periaqueductal gray, and numerous preganglionic ANS neurons. The VTA, which also receives innervation from other hypothalamic nuclei, is of particular importance. The VTA is a dopaminergic component of the mesolimbic reward pathway, which also includes the nucleus accumbens (NAc) and prefrontal cortex (PFC). The mesolimbic reward pathway is implicated in the pleasurable aspects of food consumption. Orexin and MCH from the LH serve complementary roles in the generation of behaviors involved in food consumption; orexin contributes to the initiation of food consumption, and MCH sustains food consumption once initiated. POMC neurons inhibit orexin neurons in the LH, preventing their orexigenic influences on food consumption. NPY/AgRP neurons excite orexin neurons in the LH, promoting food consumption. Like NPY/AgRP neuron signaling to the PVN, NPY/AgRP neuron signaling to the LH alone is able to induce food consumption. Lesion studies in the LH demonstrate the complexity of feeding behaviors by revealing that different processes appear to underlie the generation of nutritive feeding to maintain energy balance and the generation of hedonic feeding (i.e., eating for pleasure). Destruction of dopaminergic fibers in the LH prevents food seeking behavior without decreasing the pleasure derived from eating. In contrast, stimulation of these dopaminergic fibers tends to promote food-seeking behaviors, though also without altering the pleasure derived from eating. This demonstrates that the palatability of food is attributable to a different circuit than those underlying hunger and food drive.
Finally, POMC neurons and NPY/AgRP neurons innervate the DMH. Despite its innervation of the PVN and LH, the DMH has less salient roles on regulating food consumption. However, the DMH plays an important role in maintaining energy homeostasis by increasing energy expenditure through activation of the raphe pallidus in the brainstem, which projects serotonergic fibers that activate preganglionic sympathetic neurons in the intermediolateral nucleus of the upper thoracic spinal cord, thereby promoting energy expenditure through activation of sympathetic efferents.
End WG12
While arcuate POMC and NPY/AgRP neurons predominantly form local connections within the hypothalamus, arcuate POMC and NPY/AgRP neurons also innervate extrahypothalamic structures involved in the maintenance of energy homeostasis. NPY/ AgRP neurons release GABA at extrahypothalamic sites, including the parabrachial nucleus (PBN). NPY/AgRP neurons’ GABAergic inhibition of the PBN contributes to the promotion of food consumption. However, in contrast to NPY/AgRP signaling to the PVN or LH, NPY/AgRP neuron signaling to the PBN alone is not sufficient to induce feeding. Additionally, NPY/AgRP neurons innervate the bed nucleus of the stria terminalis (BNST), a forebrain structure involved in controlling neuroendocrine and autonomic responses, including feeding and reward behavior. The BNST connects hypothalamic nuclei to other limbic nuclei, as well as to brainstem nuclei.
In addition to responding to hormonal orexigenic and anorexigenic signals, hypothalamic neurons (and in some cases POMC neurons and NPY/AgRP neurons in the ARC) and other brain regions involved in energy homeostasis respond to neuronal inputs, including autonomic afferents that provide information on internal peripheral conditions, and neuronal inputs that convey sensory stimuli.
Autonomic afferents contribute to orexigenic and anorexigenic signaling both by detecting the presence of peripheral orexigenic and anorexigenic hormones and by sensing digestive processes. For example, in the medulla, the nucleus of the solitary tract (NTS) is involved in regulating food consumption and energy expenditure, and receives innervation from autonomic afferents. In this regard, a sub-nucleus of the NTS, the gustatory nucleus, is important. Vagus nerve afferents detect and convey the presence of certain peripheral orexigenic and anorexigenic hormones to the gustatory nucleus of the NTS. Furthermore, vagus nerve afferents also provide the gustatory nucleus with information regarding digestive processes. For example, activation of stretch receptors on the stomach during periods of gastric distention following consumption indicate the presence of large amounts of food in the stomach. This information is conveyed to the gustatory nucleus, which responds by promoting satiety through its connections to central neural circuits. The gustatory nucleus of the NTS also receives taste information due to innervation by the facial nerve via the Chorda tympani (providing taste information from the anterior two-thirds of the tongue), from the glossopharyngeal nerve (providing taste information from the posterior third of the tongue), and from the vagus nerve (providing taste information from a small portion of the epiglottis). Furthermore, the NTS itself is sensitive to certain orexigenic and anorexigenic signals, the detection of which activates appropriate appetite-promoting or appetite-suppressing responses. The NTS innervates hypothalamic nuclei, such as the PVN, and contributes to autonomic outputs involved in energy homeostasis.
The importance of external sensory stimuli in activating orexigenic and anorexigenic signaling pathways is apparent at the level of arcuate POMC neurons and NPY/AgRP neurons. During a period of fasting, visual and olfactory (i.e., smell) food stimuli prior to food consumption are sufficient to reverse the effects of fasting on POMC and NPY/ AgRP neuron activity; the fasting state-induced decreases in POMC neuron activity and increases in NPY/AgRP neuron activity are lost, yielding an anorexigenic effect. In addition to the central anticipatory anorexigenic effects of food exposure (but not consumption), the perception of impending food consumption, digestion, and absorption can stimulate the release of short-term, peripheral anorexigenic hormones through efferent autonomic signaling pathways.
Collectively, multiple brain regions, the hypothalamus in particular, integrate hormonal and neural signals. Through connections to decision making centers (e.g., the PFC, hippocampus, and amygdala), integrated signals regulate to the generation of homeostatic food drive and energy expenditure (i.e., to maintain energy homeostasis; predominantly based on signal integration by hypothalamic circuits) and hedonic food drive (involving the mesolimbic reward system). While the interactions between components of the reward system involved in food-related rewards have been primarily studied in rodents, these interactions are far more complex in humans, in which behavioral drives based on rewards are more strongly impacted by cognition.
Numerous signal molecules, both hormones and neurotransmitters, collectively reflect energy balance within the body. It is important to remember that these hormones and neurotransmitters have different effects at different synapses and tissues. For example, while it may be a potent factor promoting food consumption, NPY can also suppress ovulation and inhibit sexual behavior.
End WG13
Orexigenic Signals
Ghrelin is an orexigenic signal released during periods of fasting from the stomach and parts of the duodenum. Ghrelin promotes food consumption and decreases energy expenditure by activating arcuate NPY/AgRP neurons and LH orexin neurons. Furthermore, ghrelin inhibits PVN neurons, which prevents PVN-mediated anorexigenic signaling and energy expenditure. As a consequence, ghrelin signaling promotes adiposity. Contributing to its orexigenic effects, ghrelin also inhibits vagus nerve afferents projecting to the NTS that would otherwise promote satiety. Ghrelin is also released within the brain as a neurotransmitter.
Ghrelin signaling is reduced in obesity, likely as an adaptive response to obesity in an attempt to reduce further food consumption. In obesity, there are reduced levels of circulating ghrelin and an impaired ability of ghrelin to induce food intake through interactions with arcuate NPY/AgRP neurons. Nevertheless, stomach stapling, which removes the source of ghrelin, is a treatment for obesity. Anorexia nervosa is associated with low levels of ghrelin. In addition to its effects on feeding behavior, ghrelin also stimulates the release of growth hormones.
Glucose itself serves as a direct signal of immediate energy availability. Sufficient circulating levels of glucose exert an anorexigenic effect. Glucose inhibits orexigenic signaling by arcuate NPY/AgRP neurons and by LH. However, low levels of glucose (i.e., hypoglycemia) exert an orexigenic effect by releasing inhibition on NPY/AgRP neurons and LH neurons, permitting increased orexigenic signaling. Glucose-sensitive LH neurons co-express orexin and glutamate. When hypoglycemia releases glucose- mediated inhibition of these orexin-glutamate LH neurons, they excite dopaminergic neurons in the VTA, which promotes food consumption.
End WG14
Long-Term Anorexigenic Signals
Leptin is released from white adipocytes in proportion to their triglyceride reserves such that higher levels of leptin indicate high amounts of energy available; leptin is the most important long-term anorexigenic signal. Leptin expression is also induced by insulin.
Leptin acts directly on neurons of the hypothalamus to suppress appetite and enhance energy expenditure. In contrast, the absence of leptin is a signal that energy availability is depleted, and that consumption is required to replenish reserves. A lack of leptin (or impaired leptin signaling) permits increased appetite and food consumption while also reducing energy expenditure. Leptin-signaling-deficient humans crave food and have a slower metabolism, which can precipitate obesity. This can be due to mutations in the
gene for leptin, but can also be due to a decreased sensitivity to leptin stemming from an under-expression of hypothalamic leptin receptors, insensitive leptin receptors, or dysregulation of intracellular signaling pathways activated by leptin.
In the ARC, leptin excites POMC neurons and inhibits NPY/AgRP neurons. The leptin receptor is coupled janus kinase 2 (Jak2). Upon leptin binding, leptin receptors dimerize, enabling each Jak2 to phosphorylate the other, resulting in the activation of each Jak2. Activated Jak2 initiates numerous signaling pathways within POMC neurons, two of which are of particular importance. Firstly, activated Jak2 phosphorylates signal transducer and activator of transcription 3 (STAT3).
Phosphorylated STAT3 proteins dimerize and translocate to the nucleus. Among other effects on gene transcription, phosphorylated STAT3 dimers bind to the POMC gene promoter, increasing POMC transcription. In addition, STAT3 dimers increase expression of suppressor of cytokine signaling 3 (SOCS3), which inhibits phosphorylation/activation of Jak2, thereby providing negative feedback on the Jak2- STAT3 pathway. Secondly, Jak2 phosphorylates insulin receptor substrate proteins 1 and particularly 2 (IRS-1 and IRS-2), which then bind to and activate phosphatidylinositide-3 kinase (PI3K). PI3K generates phosphatidylinositol-3,4,5- triphosphate (PIP3), which contributes to the activation of phospholipase C γ (PLCγ) and translocation of PLCγ to the plasma membrane. Activated PLCγ at the plasma membrane hydrolyses phosphatidylinositol 4,5-biphosphate (PIP2). Among effects on other transmembrane channels, this hydrolysis activates transient receptor potential 5 (TRPC5), a type of cation channel that permits a robust inward cation current that depolarizes the membrane. The hydrolysis also yields inositol triphosphate (IP3) and diacylglycerol (DAG). IP3 liberates intracellular calcium stores by binding to IP3 receptors on the endoplasmic reticulum. Additionally, PI3K accelerates the translocation and insertion of TRPC5 channels into the POMC neuronal membrane. Thus, through activation of STAT3 pathways, leptin increases POMC transcription, which ultimately increases the release of anorexigenic melanocortins, and through activation of IRS- PI3K pathways, leptin excites POMC neurons by increasing translocation and activation of TRPC5, which increases membrane depolarization. In addition to exciting POMC neurons, leptin hyperpolarizes (i.e., inhibits) NYP/AgRP neurons through PI3K-mediated activation of ATP-sensitive potassium channels (i.e., they open ATP-sensitive potassium channels, permitting potassium efflux). Furthermore, through the leptin receptor- activated STAT3 pathway, leptin inhibits expression of NPY and AgRP.
Both orexin and MCH levels are inversely related to leptin levels, largely mediated by leptin-responsive arcuate POMC and NPY/AgRP neurons that innervate orexin and MCH neurons in the LH. In other words, high leptin leads to inhibition of LH orexin and MCH neurons. Leptin also has important excitatory effects on VMH neurons.
Since leptin serves as a continuous signal of energy availability, leptin exerts a tonic excitatory effect on POMC and VMH neurons and a tonic inhibitory effect on NPY/AgRP neurons. This means that if leptin signaling is somehow impaired, there will be decreased activation of POMC and VMH neurons and a release of inhibition on NPY/ AgRP neurons, increasing NPY/AgRP signaling.
In addition to activating leptin receptors in hypothalamic neurons, leptin also activates receptors on dopaminergic VTA neurons of the mesolimbic reward system, which reduces hedonic feeding and behavioral drive for high-sugar and high-fat foods.
End WG15
Short-Term Anorexigenic Signals
Insulin serves as a short-term anorexigenic signal through direct interactions with central receptors, which results in reduced food consumption and increased energy expenditure. However, insulin also indirectly promotes hunger by decreasing blood glucose, low levels of which release inhibition on orexigenic signaling by arcuate NPY/ AgRP neurons and LH orexin neurons, enabling their orexigenic signaling pathways to stimulate food consumption and reduce energy expenditure. In fact, this indirect appetite-promoting effect of insulin contributes to the initiation of food consumption. In anticipation of food, vagus nerve efferents stimulate insulin secretion by β cells. This causes a slight decrease in blood glucose levels. The decreased blood glucose releases glucose-mediated inhibition of arcuate NPY/AgRP neurons and some glucose- sensitive LH orexin neurons, promoting food consumption. Once food consumption is initiated, insulin secretion continues to increase to accommodate rising levels of blood glucose as food is absorbed. The rising blood glucose promotes satiety as it reaches concentrations sufficient to inhibit NPY/AgRP neurons and glucose-sensitive LH orexin neurons.
The direct anorexigenic effects of insulin receptor activation are very similar to those of leptin receptor activation; both operate on most of the same brain regions, and even share common downstream intracellular signaling pathways. Like leptin receptor activation in POMC neurons, insulin receptor activation on POMC neurons initiates an IRS-PI3K, which depolarizes and increases the firing rates of POMC neurons by increasing recruitment and activation of TRPC5. Upon insulin binding, insulin receptors dimerize, activating insulin receptor tyrosine kinase. Insulin receptor tyrosine kinase phosphorylates and recruits substrate proteins, including IRS-1 and IRS-2.
Phosphorylated IRS-1 and IRS-2 activate PI3K, which generates PIP3. PIP3 contributes to the activation of PLCγ and translocation of PLCγ to the plasma membrane. PIP3 also increases recruitment of TRPC5. Activated PLCγ at the plasma membrane is then able to hydrolyze PIP2. The hydrolysis of PIP2 activates TRPC5 channels, which increase cation conductance and depolarize the membrane. While both leptin receptor activation and insulin receptor activation on POMC neurons are required for efficient anorexigenic signaling, leptin and insulin affect different subpopulations of arcuate POMC neurons.
Like leptin, insulin also hyperpolarizes NPY/AgRP neurons through PI3K-mediated activation of ATP-sensitive potassium channels. However, unlike leptin, insulin receptor activation on NPY/AgRP neurons has a very important role in glucose homeostasis.
Insulin receptor activation on NPY/AgRP neurons is required for insulin to suppress hepatic glucose production. Insulin-mediated inhibition of NPY/AgRP neurons affects hepatic innervation, leading to increased expression of interleukin 6 (IL-6) in hepatic
parenchymal cells. In hepatic parenchymal cells, IL-6 decreases the expression of glucose-6-phosphatase, reducing gluconeogenesis.
Leptin and insulin both excite the VMH by targeting VMH-specific steroidogenic factor 1 (SF-1) neurons. VMH-specific SF-1 neurons excite arcuate POMC neurons through glutamatergic synapses, promoting satiety and energy expenditure. However, when induced by a high-fat diet, insulin-mediated activation of VMH-specific SF-1 neurons contributes to the development of obesity. Deletion of insulin receptors in these VMH- specific SF-1 neurons results in increased POMC neuron activity under high-fat diet conditions, and prevents diet-induced obesity and diet-induced impairments in glucose metabolism. In contrast, increased leptin signaling in VMH SF-1 neurons improves glucose homeostasis without affecting body weight.
Like leptin, insulin receptor activation on dopaminergic neurons of the mesolimbic reward system reduces hedonic feeding and behavioral drive for high-sugar and high-fat foods. However, the effects of insulin receptor activation on dopamine release is dependent on nutritional context. Insulin stimulates dopamine release under food- restricted conditions, but not under obesogenic conditions.
Since insulin is both a direct satiety signal and an indirect satiety signal (through its hypoglycemic effects), anything that stimulates or enhances insulin release will indirectly promote satiety.
End WG16
Cholecystokinin (CCK) is secreted by I cells in the duodenum and jejunum in response to the presence of fatty acids in the small intestine or in response to cholinergic innervation by vagus nerve efferents. CCK is also used as a neurotransmitter by enteric nervous system neurons as well as in the brain. CCK receptors are found throughout the brain, but circulating CCK is unable to permeate the blood-brain barrier. As a consequence, peripheral CCK is only able to exert anorexigenic effects through interactions with vagus nerve afferents and in brainstem nuclei and hypothalamic regions lacking a normal blood-brain barrier. Peripheral CCK stimulates anorexigenic vagus nerve afferents projecting to the NTS, which decreases meal frequency and size. This effect of CCK directly opposes the effects of ghrelin mediated by vagus nerve afferents. In addition to vagus nerve-mediated anorexigenic effects on the NTS, CCK also interacts with the NTS directly. Circulating CCK also exerts anorexigenic effects through interactions with the ARC, PVN, VMH, DMH, and LH.
End WG17
Glucagon-like peptide 1 (GLP-1) is secreted by intestinal L cells, found from the duodenum to the colon in increasing concentrations (i.e., they are most concentrated in the ileum), in response to nutrients in the small intestine. GLP-1 is also used as a neurotransmitter in the central nervous system, where it is expressed by neurons of the NTS and the reticular nucleus of the medulla. Central GLP-1 influences energy homeostasis at multiple levels within the central nervous system. The GLP-1 receptor is expressed in regions distributed throughout the brain, including in the circumventricular organs, the amygdala, the ventrolateral medulla, the NTS, the thalamic paraventricular
nucleus, the hippocampus, the cortex, the PBN, and the hypothalamus (particularly in hypothalamic regions involved in energy homeostasis).
Circulating GLP-1 serves as an incretin: it enhances glucose-dependent insulin secretion by pancreatic β cells. Thus, GLP-1 exerts indirect anorexigenic effects by enhancing anorexigenic insulin signaling. However, circulating GLP-1 also has anorexigenic effects itself through stimulation of vagus nerve afferents projecting to the NTS. Furthermore, circulating GLP-1 is able to permeate the blood-brain barrier, which enables activation of receptors throughout the brain.
GLP-1 promotes satiety and increases energy expenditure through interactions with receptors found in the ARC, VMH, DMH, NTS, and PBN. GLP-1 also exerts direct actions on the VTA and NAc of the mesolimbic reward system, which reduces food reward-related behaviors, selectively reducing high-fat food consumption while increasing low-fat food consumption (when a choice between types of food is available). Beyond its central roles in the regulation of energy balance, central GLP-1 also appears to serve neuroprotective and neurogenic roles.
End WG18
In addition to releasing GLP-1, intestinal L cells also secrete peptide YY (PYY) in response to nutrients in the small intestine. Like GLP-1, PYY is used as a neurotransmitter by the reticular nucleus of the medulla. PYY promotes satiety through stimulation of vagus nerve afferents projecting to the NTS, while also exerting anorexigenic effects through direct interactions with the NTS, as well as through direct interactions with the ARC, PVN, VMH, and DMH. In the ARC, PYY inhibits NPY/AgRP neurons. NPY/AgRP neurons express Y2Rs, inhibitory NPY autoreceptors. PYY inhibits NPY/AgRP neurons by activating these inhibitory Y2Rs (i.e., PYY is an agonist at Y2Rs).
Peripheral amylin, co-secreted with insulin by pancreatic β cells, can cross the blood- brain barrier to stimulate amylin receptors, which are distributed throughout the brain, with concentrations in regions involved in the control of energy homeostasis. The anorexigenic effects of amylin compliment those of insulin. The amylin receptor- expressing regions involved in energy homeostasis include the VTA and NAc of the mesolimbic reward system, the hypothalamus, caudal brainstem nuclei involved in feeding behavior, the lateral PBN, and especially the area postrema. Amylin receptor
activation in these regions promotes satiety and increases energy expenditure. Amylin appears to exert its most salient anorexigenic effects through interactions with glucose- sensitive neurons in the area postrema, which are inhibited in the presence of low glucose. However, amylin receptor activation in the hypothalamus also plays critical roles in amylin’s effects on energy homeostasis.
In addition to peripheral secretion of amylin, amylin is also released as a neurotransmitter by the ARC, the preoptic area of the hypothalamus, and the LH. Amylinergic LH neurons are innervated by amylin-sensitive lateral PBN neurons, indicating that peripheral amylin secretion may promote central amylinergic signaling.
Within the mesolimbic reward system, amylin induces food consumption-suppressing effects and body weight-suppressing effects. Amylin inhibits dopaminergic signaling by the VTA to the NAc core (NAcC). In the long term, amylin-mediated suppression of food consumption through inhibition of dopaminergic VTA-NAcC signaling appears to favor suppression of palatable food consumption over suppression of bland food consumption. Amylin binds robustly to neurons in the NAc itself, wherein amylin interacting with the NAc shell (NAcSh) may suppress µ-opioid receptor-induced hyperphagia. However, unlike the ability of amylin to independently suppress food consumption through interactions with the area postrema and VTA, amylin may not be able to independently suppress food consumption through interactions with the NAcSh. Rather, amylin may only suppress food consumption through modulation of, or interaction with, opioid signaling pathways.
This modulatory role of amylin is evident in anorexigenic signaling pathways. Amylin seems to enhance anorexigenic signaling by other satiety hormones, such as leptin, insulin, CCK, and PYY, when amylin is co-administered. Most notably, amylin interacts cooperatively with leptin to suppress appetite and increase energy expenditure, resulting in the selective reduction of adiposity without reducing lean mass. The anorexigenic effects of leptin paired with amylin are greater than the sum of leptin and amylin’s independent anorexigenic effects. In other words, there is a greater-than- additive effect on satiety when leptin and amylin signaling pathways are simultaneously activated. The VMH and ARC are sites at which amylin-leptin interactions enhance appetite suppression and energy expenditure. In both the VMH and the ARC, the administration of amylin increases expression of the leptin receptor, enhances leptin binding, and increases leptin-induced phosphorylation of STAT3. In the VMH, activation of amylin receptors on microglia (resident central nervous system macrophages) induces microglial production of IL-6. IL-6 activates IL-6/gp130 receptor complexes on leptin-sensitive VMH neurons, which activates STAT3. Amylin-enhanced leptin signaling requires amylin-mediated induction of IL-6. STAT3 is phosphorylated downstream of both amylin and leptin receptor activation. Phosphorylated STAT3 proteins dimerize, translocate to the nucleus, and regulate gene transcription.
Beyond its effects on energy homeostasis, amylin at high concentrations also seems to exert cognitive enhancing effects, and exogenous administration of amylin may mitigate
neurodegenerative and cognitive effects of Alzheimer’s disease. However, whether this is achieved through direct interactions in the central nervous system is unclear.
In addition to hormones involved in regulating energy homeostasis, certain neurotransmitter systems are also implicated. For example, serotonin generally has anorexigenic effects; serotonin levels rise in anticipation of food, spike while eating (especially while eating carbohydrate-rich foods), and then fall as food is absorbed. The appetite-suppressing effects of serotonin are supported by studies demonstrating how drugs that enhance serotonergic signaling tend to be anorexigenic. In addition to its proposed role in mood disorders, impaired serotonergic signaling is implicated in hypophagic eating disorders, particularly bulimia nervosa.
End WG01
Neuroendocrinology of Energy Expenditure: Adaptive Thermogenesis
Maintenance of energy homeostasis relies not only upon the efficient regulation of food consumption, but also on the efficient regulation of energy expenditure. As such, most appetite-promoting and appetite-suppressing signals also affect energy expenditure.
One of the primary ways in which energy expenditure is regulated is through the promotion or suppression of adaptive thermogenesis (also called non-shivering thermogenesis), a process in which chemical energy reserves are dissipated as heat in specialized adipose tissue.
Based on integrated signals of energy availability, energy homeostasis-maintaining pathways can activate adaptive thermogenesis to expend excess energy reserves when indicated by anorexigenic signals or can inhibit adaptive thermogenesis to conserve depleted energy reserves when indicated by orexigenic signals. By regulating adaptive thermogenesis, the hypothalamus can compensate for the often-imperfect balance between energy consumption and energy expenditure. However, adaptive thermogenesis also permits efficient maintenance of body temperature within a discrete range when controlled by thermoregulatory pathways.
There is substantial overlap between energy homeostasis-maintaining pathways and thermoregulatory pathways controlling adaptive thermogenesis. Furthermore, adaptive thermogenesis also serves as a component of immune responses by contributing to fever during infection. Like the circuits responsible for the maintenance of energy homeostasis, circuits responsible for thermoregulation are predominantly located within the hypothalamus. Outputs governing adaptive thermogenesis from central energy homeostasis-maintaining circuits and central thermoregulatory circuits activate sympathetic efferents that innervating adipose tissue, where adaptive thermogenesis occurs.
End WG02
There are two basic types of adipose tissue, white adipose tissue and brown adipose tissue. White adipose tissue and brown adipose tissue are distributed in different regions within the body and exhibit distinct profiles of gene expression that result in distinct morphological and functional features. White adipose tissue (WAT) lacks thermogenic capacity and primarily serves as a reservoir for energy stored as triglycerides. White adipocytes contain very few mitochondria and a single, large lipid droplet. WAT is most commonly distributed as subcutaneous tissue around the abdomen, waist, and thighs. Brown adipose tissue (BAT) has substantial thermogenic capacity; adaptive thermogenesis is the primary function of BAT. BAT is distributed in subscapular, cervical, peri-spinal, mediastinal, periaortic, pericardial, and periadrenal regions. The BAT in these regions arises myogenic-factor 5-expressing cells, which are also myocyte precursors. Unlike the single large lipid droplet and limited mitochondria present in each white adipocyte, brown adipocytes contain plentiful small lipid droplets surrounded by numerous mitochondria. Iron-pigmented cytochromes in the mitochondria function as electron transport agents while also causing the brown coloration of BAT. Furthermore, there is increased vascularization of BAT compared to WAT, which supports oxygen and nutrient exchange necessary for the high metabolic demands of BAT while also facilitating the distribution of BAT-generated heat throughout the body. These features enable the utilization of large amounts of energy for thermogenesis.
End WG03
Adaptive thermogenesis requires the presence of uncoupling protein 1 (UCP1, also called thermogenin), a protein found in the inner membranes of brown adipocyte mitochondria. UCP1 is a long-chain fatty acid (LCFA) anion/proton symporter, but because hydrophobic interactions with hydrophobic LCFA tails prevent LCFA dissociation, UCP1 functions as an LCFA-activated proton channel, permitting mitochondrial proton leakage. The result is that UCP1 activation by LCFAs uncouples substrate oxidation from electron transport, dissipating oxidation energy as heat. UCP1- mediated thermogenesis is inhibited by nucleotides.
Brown adipocytes express a high concentration of β3 adrenergic receptors and receive rich innervation from noradrenergic sympathetic fibers, which are regulated by hypothalamic energy homeostasis-maintaining circuits and hypothalamic thermoregulatory circuits. Sympathetic activation of β3 adrenergic receptors in adipocytes is required for the initiation of lipolysis and thermogenesis. BAT receives sympathetic innervation from sympathetic ganglia, which are activated by preganglionic fibers in the intermediolateral nucleus of the upper thoracic spinal cord.
β3 adrenergic receptors are GS-protein-coupled receptors, and are primarily involved in stimulating adaptive thermogenesis in adipose tissue. β3 receptor activation in BAT activates adenylyl cyclase, which increases production of cAMP. Accumulating cAMP activates protein kinase A (PKA), which phosphorylates multiple lipases, including triacylglycerol lipase, diacylglycerol lipase, and monoacylglycerol lipase. Through lipolysis of triglycerides, lipases generate LCFAs, which both activate UCP1 and serve as energy substrates. PKA also activates p38 mitogen-activated protein kinase (MAPK)
and extracellular signal regulated kinase 1 and 2 (ERK1/2), which increase expression of UCP1. β3 receptor activation also increases the quantity of mitochondria per adipocyte and promotes the expression of other thermogenic proteins. Collectively, β3 receptor activation increases BAT thermogenic capacity.
Thermoregulatory circuits in the hypothalamus activate sympathetic efferents innervating adipose tissue when those circuits receive sufficient signals of cold exposure. Cold-sensitive neurons are activated when temperatures fall below a set point. Activation of cold-sensitive neurons projecting to thermoregulatory circuits will lead to activation of thermoeffectors that conserve and generate heat, the most important of which are cutaneous vasoconstriction (preventing heat loss from the surface of the skin), shivering, and adaptive thermogenesis in BAT, but which also include the induction of goose bumps and warmth-seeking behaviors. Warm-sensitive neurons are activated by temperatures above a set point. Activation of warm-sensitive neurons initiates thermoregulatory signaling pathways that result in the shunting of blood towards the periphery to dissipate heat, the stimulation of sweating to further dissipate heat through evaporative cooling, and the initiation of shade-seeking behaviors. Furthermore, warm-sensitive neuron activation will result in inhibition of pathways promoting adaptive thermogenesis to prevent further heat generation.
Efficient thermoregulation through adaptive thermogenesis in BAT begins with efficient integration of peripheral temperature signals. Cutaneous transient receptor potential (TRP) cation channels on temperature-sensitive primary sensory neurons respond to changes in peripheral body temperature. Cold-sensitive primary sensory neurons express TRP cation channels activated by cold (e.g., TRPM8, also called cold and menthol receptor 1, CMR1). As the temperature to which a cold-sensitive neuron is exposed decreases, more TRP cation channels on that cold-sensitive neurons will be activated, increasing cation conductance and consequent membrane depolarization. At a certain point (i.e., that cold-sensitive neuron’s set point), cation conductance through activated TRP channels will permit sufficient depolarization to generate an action potential in the cold-sensitive neuron. Warm-sensitive primary sensory neurons express TRP cation channels activated by heat (e.g., TRPV1). As the temperature to which a warm-sensitive neuron is exposed increases, more TRP cation channels on that warm- sensitive neuron will be activated, increasing cation conductance and consequent membrane depolarization. At a certain point, cation conductance through activated TRP channels will permit sufficient depolarization to generate an action potential.
Temperature-sensitive primary sensory neurons have cell bodies located in dorsal root ganglia. They are unipolar neurons, each projecting a single process from its cell body, which extends from cutaneous dendrites to spinal axon terminals. A temperature- sensitive primary sensory neuron enter the spinal cord through a dorsal root, wherein its cell bodies resides, but extends beyond its cell body into the dorsal horn, where its axon terminals innervate a glutamatergic second-order neuron. The second-order neuron projects to the lateral PBN (LPBN). Cold-sensitive dorsal horn neurons project to glutamatergic third-order neurons in the lateral sub-nucleus of the LPBN, and warm- sensitive dorsal horn neurons project to glutamatergic third-order neurons in the dorsal
sub-nucleus of the LPBN. Glutamatergic third-order temperature-sensitive neurons of the LPBN innervate the median preoptic nucleus (MnPO) of the hypothalamus, a sub- nucleus of the preoptic area (POA) of the hypothalamus.
The POA is the primary thermoregulatory center of the brain. In the POA, cutaneous temperature signals and core temperature signals are integrated to generate appropriate responses by thermoeffectors. The POA is divided into sub-nuclei, including the MnPO, medial preoptic nucleus (MPO), lateral preoptic nucleus (LPO), and the preoptic periventricular area (POP).
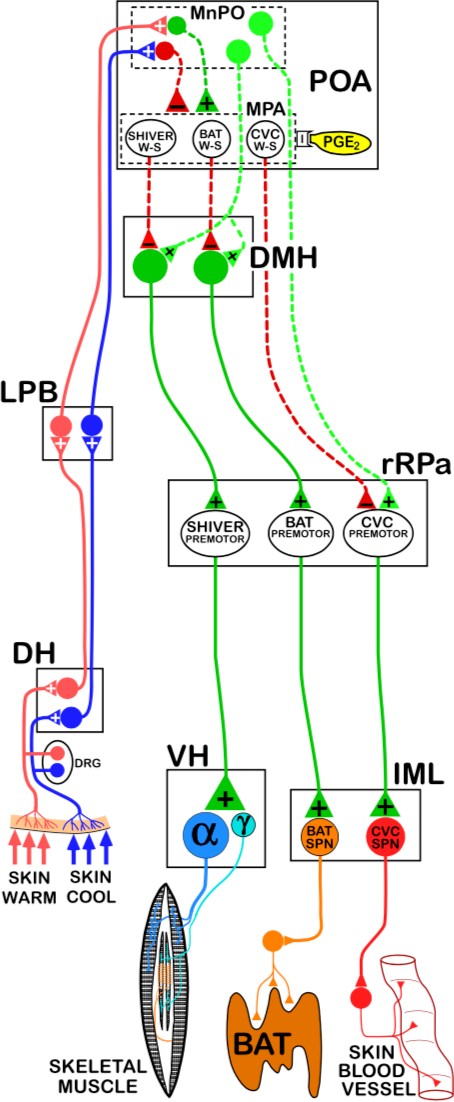
Third-order cold-sensitive glutamatergic LPBN innervate GABAergic neurons in the MnPO, and third-order warm-sensitive glutamatergic neurons in the dorsal sub-nucleus of the LPBN innervate glutamatergic neurons in the MnPO. Thus, GABAergic MnPO neurons will be activated in response to cutaneous cold exposure and glutamatergic MnPO neurons will be activated in response to cutaneous heat exposure. These GABAergic MnPO neurons and glutamatergic MnPO neurons innervate distinct populations of intrinsically warm- sensitive GABAergic neurons in the MPO.
These warm-sensitive GABAergic MPO neurons in the MPO are the convergence point for cutaneous temperature signals (relayed by MnPO neurons) and central temperature signals (directly sensed by MPO neurons). Based on integration of cutaneous temperature signals in the MnPO and central temperature signals in the MPO, the POA generates appropriate thermoregulatory responses to changing internal and external temperature conditions.
Distinct populations of intrinsically warm- sensitive GABAergic MPO neurons regulate different thermoeffector pathways, but all exert tonic inhibition upon the thermoeffector pathways with which they interact (i.e., some populations tonically inhibit pathways that activate cutaneous vasoconstriction, some populations tonically inhibit pathways that activate shivering, and some populations tonically inhibit pathways that activate adaptive thermogenesis in BAT).
Adaptive thermogenesis in BAT requires β3 receptor activation by sympathetic fibers, which are regulated by preganglionic neurons in the intermediolateral nucleus. Efferent control of adaptive thermogenesis in BAT by the POA is largely mediated by regulation of glutamatergic DMH neurons. Glutamatergic DMH neurons innervate serotonergic neurons in the rostral raphe pallidus (rRPa). These serotonergic rRPa neurons activate preganglionic neurons in the intermediolateral nucleus, which stimulate sympathetic activation of adaptive thermogenesis in BAT. Thus, activation of glutamatergic DMH neurons induces downstream activation of adaptive thermogenesis in BAT.
However, glutamatergic DMH neurons are tonically inhibited by intrinsically warm- sensitive GABAergic MPO neurons. Activation of glutamatergic DMH neurons and subsequent downstream activation of adaptive thermogenesis in BAT involves the release of tonic inhibition by GABAergic MPO neurons. While pathways activated in response to cold exposure promote the release of inhibition on glutamatergic DMH neurons (and subsequent activation of BAT thermogenesis), pathways activated in response to heat exposure enhance inhibition of glutamatergic DMH neurons. Activity of the warm-sensitive GABAergic MPO neurons is affected by local hypothalamic temperature. Central heat excites the GABAergic MPO neurons, leading to enhanced tonic inhibition. However, central cold exerts an inhibitory effect on the warm-sensitive GABAergic MPO neurons, which reduces tonic inhibition of glutamatergic DMH neuron, promoting activation of adaptive thermogenesis in BAT.
Figure WG04
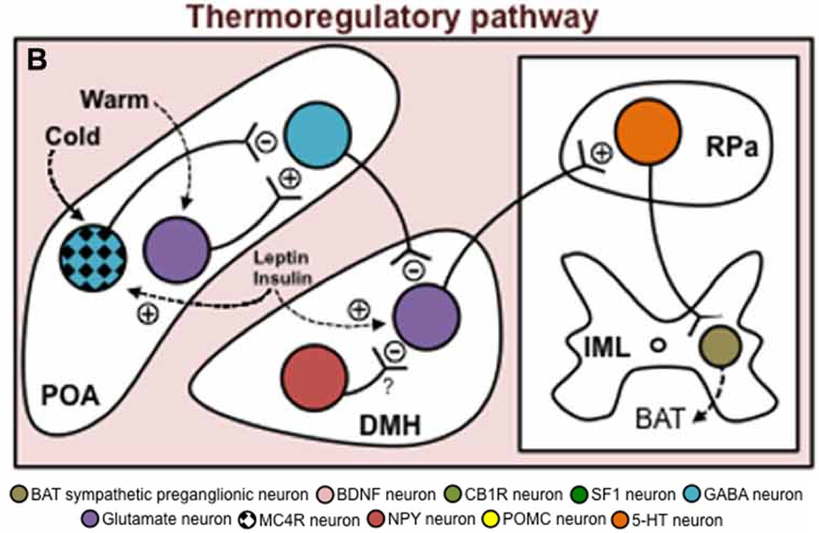
Through afferent sensory pathways, cutaneous temperature signals also affect the warm-sensitive GABAergic MPO neurons. Cutaneous heat exposure leads to activation of glutamatergic MnPO neurons, which excite warm-sensitive GABAergic MPO neurons, enhancing their tonic inhibition of glutamatergic DMH neurons and thereby
inhibiting BAT thermogenesis. Cutaneous cold exposure leads to activation of GABAergic MnPO neurons, which inhibit warm-sensitive GABAergic MPO neurons, releasing inhibition of glutamatergic DMH neurons, and promoting their activation of BAT thermogenesis.
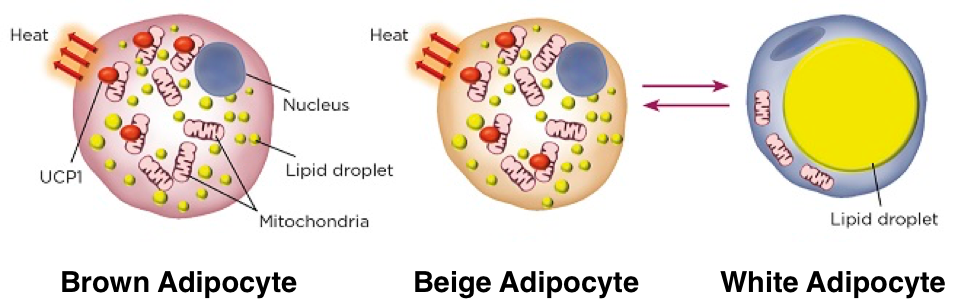
Though WAT lacks thermogenic capacity, WAT responds to most of the signals that increase thermogenesis in BAT. WAT receives significant noradrenergic innervation by sympathetic fibers (though not to the degree found in BAT). Like in BAT, activation of β3 adrenergic receptors on WAT activates adenylyl cyclase, which leads to accumulation of cAMP and subsequent activation of PKA. PKA can then phosphorylate lipases to increase lipolysis and activate intracellular signaling pathways.
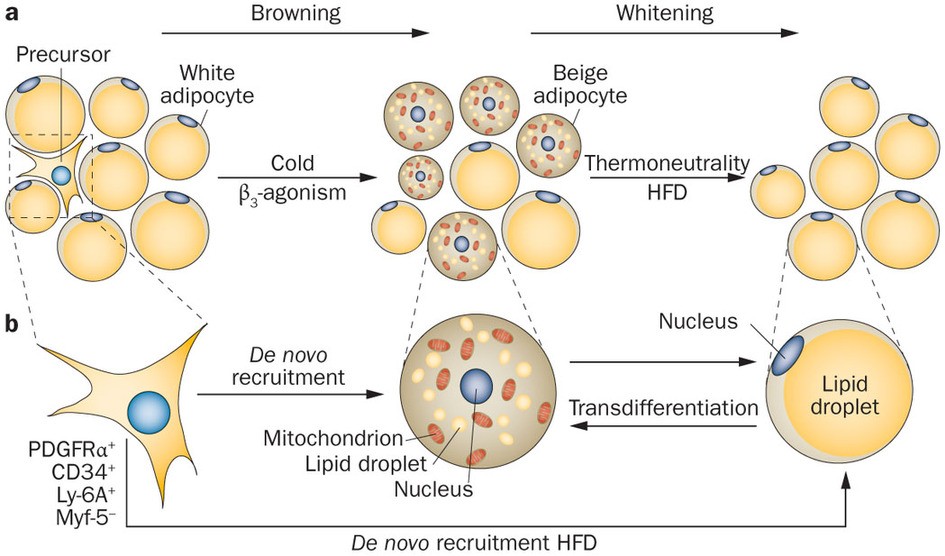
During periods of sustained sympathetic activation, due to prolonged cold exposure or direct β3 adrenergic receptor activation, some white adipocytes undergo a process of ‘browning,’ that results in increased thermogenic capacity through increased numbers of mitochondria, increased numbers of tiny lipid droplets, and most importantly, increased
expression of UCP1. The resultant brown adipocyte-like cells are called beige adipocytes. However, without persistent cold or direct β3 agonism (e.g., during a period of thermoneutrality), beige adipocytes will undergo a process of ‘whitening,’ in which they revert back to their original white adipocyte profile.
Start WG05
In addition to prolonged cold exposure or sustained direct β3 receptor activation, physical exercise also induces the browning of WAT and activation of adaptive thermogenesis in BAT. The exercise-induced increase in WAT browning and activation of BAT is largely mediated by exercise-induced expression of irisin, a peptide hormone released by white adipocytes, brown adipocytes, and myocytes. Exercise induces skeletal muscle expression of a transcriptional regulator called peroxisome proliferator-activated receptor-γ co-activator 1 α (PGC1-α). PGC1-α controls mitochondrial biogenesis and serves as a co-activator of peroxisome proliferator- activated receptor γ (PPAR-γ). In addition to regulating UCP1 expression, PPAR-γ activates expression of fibronectin type III domain-containing protein 5 (FNDC5).
Irisin results from the proteolytic cleavage of the membrane-bound FNDC5. Irisin increases UCP1 expression in adipocytes. Irisin-induced expression of UCP1 drives browning of WAT. Irisin also exerts central anorexigenic effects within the hypothalamus similar to leptin. Irisin increases POMC and CART expression in POMC neurons and inhibits orexin.
End WG05
Neuroendocrinology of Energy Homeostasis in Obesity
- Obesity is associated with chronic, low-grade inflammation in both the periphery and central nervous system, and results in dysregulated leptin and insulin signaling. The dysregulated leptin and insulin signaling are characterized by an inability of leptin and insulin to adequately signal the presence of sufficient or excessive energy availability, which results in increased appetite and decreased energy expenditure, both of which perpetuate obesity.
- Even after only a few days of a high-fat, calorie-rich diet, an increased amount of saturated fatty acids from the periphery cross the blood-brain barrier and induce a hypothalamic inflammatory response involving the activation of microglia (resident central nervous system macrophages). Hypothalamic inflammation impairs leptin and insulin signaling, resulting in dysregulated energy homeostasis due to hyperphagia, decreased energy expenditure, and impaired glucose regulation. Mediobasal hypothalamic inflammation increases endoplasmic reticulum stress in arcuate neurons, anterior PVN neurons, and at the median eminence, which prevents leptin and insulin from efficiently activating their anorexigenic signaling cascades. In NPY/AgRP neurons, constitutive activation of the proinflammatory c-Jun N-terminal kinase 1 (JNK1) pathway promotes spontaneous NPY/AgRP neuron action potentials and leads to central and peripheral insensitivity to leptin’s effects. Similarly, constitutive activation of the inflammatory inhibitor of nuclear factor κ B kinase 2 (IKK2) pathway attenuates the NPY/AgRP neuron response to insulin, impairing glucose homeostasis by preventing insulin from inducing hepatic IL-6 signaling necessary for insulin to inhibit hepatic gluconeogenesis. In other words, the hypothalamic inflammation prevents leptin- and insulin-mediated suppression NPY/AgRP neuron signaling.
- Impaired leptin and insulin signaling in the arcuate nucleus also results from increased expression of SOCS3. SOCS3 inhibits phosphorylation/activation of Jak2, thereby preventing the ability of leptin to activate intracellular signaling pathways; the signaling pathways, which would normally mediate leptins effects on POMC neurons and NPY/AgRP neurons are insensitive to the presence of leptin. SOCS3 also inhibits intracellular signaling pathways activated by insulin. SOCS3 inhibits the insulin receptor tyrosine kinase, preventing substrate phosphorylation (including IRS-1 and IRS-2). SOCS3 also competitively binds to sites on IRS-1 and IRS-2, preventing their binding to the insulin receptor, which is necessary for their phosphorylation by the insulin receptor tyrosine kinase. SOCS3 also targets IRS-1 and IRS-2 for degradation. Like leptin, the signaling pathways that would normally mediate insulin’s effects on arcuate neurons are insensitive to the presence of insulin.
- Due to the insensitivity to leptin and insulin, the dysregulated leptin signaling and dysregulated insulin signaling are commonly referred to as ‘leptin resistance’ and ‘insulin resistance.’ However, the dysregulation of leptin and insulin signaling in obesity does not always entail a loss of sensitivity.
- In contrast to arcuate POMC and NPY/AgRP neurons that develop insulin resistance following consumption of a high-fat diet, VMH-specific SF-1 neurons exhibit an increased sensitivity to insulin in response to a high-fat diet, which impairs VMH-specific SF-1 neurons’ glutamatergic activation of arcuate POMC neurons, yielding reduced POMC neuron activation.
- Leptin sensitivity in the DMH is preserved during obesity and contributes to hypertension, likely through an increase in sympathetic output.
- It is actually selective leptin resistance and selective insulin resistance because while some CNS regions are resistant to leptin and insulin, other regions have an increased sensitivity. The decreased sensitivity in some regions and increased sensitivity in other regions all contribute to hyperphagia, adiposity, and impaired glucose homeostasis.
- Even following development of selective leptin resistance (e.g., in obesity) exogenous administration of amylin can restore leptin sensitivity.
- Unlike some other anorexigenic signals, there is no PYY resistance in obesity, but there are reduced levels of PYY. Because PYY receptors remain functional in obesity, exogenous PYY administration may be a viable treatment for obesity.
EndWG 06
Neuroendocrinology of Thirst
Thirst is stimulated by a decrease in blood volume (hypovolemia) or an increase in blood solute concentration. However, the thirst generated by a decrease in blood volume, volumetric thirst, is regulated by different mechanisms than thirst arising from high osmolarity, osmometric thirst.
When there is a decrease in blood volume, the kidneys secrete renin into circulation, which leads to the production of angiotensin II. Angiotensin II stimulates the subfornical organ of the telencephalon, which in turn activates vasopressin release by magnocellular neurons of the hypothalamus. A decrease in blood pressure is also sensed by baroreceptors on blood vessels, which signal the release of vasopressin via the vagus nerve and nucleus of the solitary tract. The release of vasopressin by the hypothalamus is a humoral response to prevent further decreases in blood pressure by increasing retention of water in the kidneys. However, the loss of blood volume also stimulates volumetric thirst and subsequent water-seeking behaviors, processes most likely involving the LH.
The OVLT (organum vascularum lateral terminalis) lacks a blood- brain-barrier, allowing neurons in the OVLT to fire proportionally to the concentration of solutes in the blood. This allows OVLT neurons to directly stimulate the release of VP from magnocellular neurons, as well as to generate osmometric thirst. While thirst and VP release are generally related, loss of VP-releasing magnocellular neurons, or the loss of functional VP receptors in the kidneys, results in diabetes insipidus, characterized by unquenchable thirst because any water consumed is quickly excreted by the renal system.
In addition to its production in gastrointestinal S cells, secretin is also produced by magnocellular neurons in both the paraventricular nucleus of the hypothalamus and in the supraoptic nucleus of the hypothalamus. Secretin in these magnocellular neurons is released from the posterior pituitary in response to increased osmolarity.
Feedback/Errata