
4 Endocrine Regulation of Energy Homeostasis
Endocrine Regulation of Energy Homeostasis
Introduction
Consumed food is stored as energy in the form of two primary macromolecules, glycogen and triglycerides, both of which are created through anabolic processes. Glycogen reserves, stored in the liver and skeletal muscles, have a finite capacity. In contrast, triglyceride reserves in adipose tissue have an almost unlimited capacity. Between periods of energy consumption, glycogen and triglycerides are catabolized to release a continuous supply of glucose for use by the brain, as well as glucose, ketones, and fatty acids for the rest of the body. For energy reserves to remain in balance, energy consumption must ultimately equal energy expenditure. When energy consumption exceeds expenditure, adiposity will increase. If energy consumption fails to meet energy demands, starvation will result. Multiple mechanisms collectively create a balance between energy consumption and energy expenditure (in both the short term and long term). Some signaling pathways influence the digestion, absorption, and storage of consumed energy sources; other signaling pathways influence behaviors driving consumption; and some signaling pathways modulate internal metabolic processes that utilize energy. There is substantial overlap in signaling pathways involved in various components of energy homeostasis. Here, we will focus primarily on those signaling pathways that influence digestion, absorption, and storage of consumed energy sources, and some of the signaling pathways that regulate energy consumption.
Regulation of digestion, absorption, and storage of consumed energy is largely achieved through gastric hormone signaling pathways. Different types of food entering the gastrointestinal tract stimulate secretions of different hormones, which signal the presence of those different types of food and generate appropriate responses. Some signals modulate digestive processes by regulating the excretion of enzymes into the digestive tract, or by changing the rate at which ingested food is transported through the digestive tract. These gastric hormones often operate at the level of the brain as well (particularly in the hypothalamus), where they regulate behaviors involved in energy consumption. Before discussing these gastric hormones, it is important to understand the basic cycle of energy consumption.
Energy Consumption
Fasting generally refers to any time in which food is not passing through the alimentary canal. The process of consuming food can be broken down into multiple stages. The first is the cephalic phase, in which feeding is anticipated through sight, smell, or conscious thoughts of food. This activates the parasympathetic and enteric nervous systems, which prepare the body for impending digestion. This preparation for impending digestion involves secretion of hormones. The cephalic phase is followed by the gastric phase, during which consumed food is digested. The digestive process itself also initiates hormone signaling pathways. The process of consuming food ends with the absorption of food, called the substrate phase. Throughout this cycle of eating and fasting, numerous neural and endocrine systems are involved in regulating appetite, digestion, absorption, storage, and usage of nutrients.
Certain regions of the brain, particularly the hypothalamus, respond to signals of energy availability within the body and contribute to feeding behaviors that replenish their availability. Orexigenic signals are those that drive feeding behaviors. These signals are often suppressed by satiety (anorexigenic) signals, which have appetite reducing effects. Orexigenic signals are produced in response to low energy availability. Orexigenic signals gradually increase as the newly consumed energy stores are expended. Over time, the rise in orexigenic signaling is accompanied by a decrease in anorexigenic signaling, ultimately leading to initiation of another meal. Anorexigenic signals are produced during digestion and from energy reserves to signal adequate energy availability. However, different orexigenic and anorexigenic signals operate over different timelines, and govern distinct aspects of feeding behavior and energy utilization.
Gastric Hormones
To begin, we will assume that orexigenic signaling prevails, and food is consumed, after which it enters the stomach.
Release of Gastrin
The presence of protein-rich foods accumulating in the lower stomach or stimulation by vagus nerve affer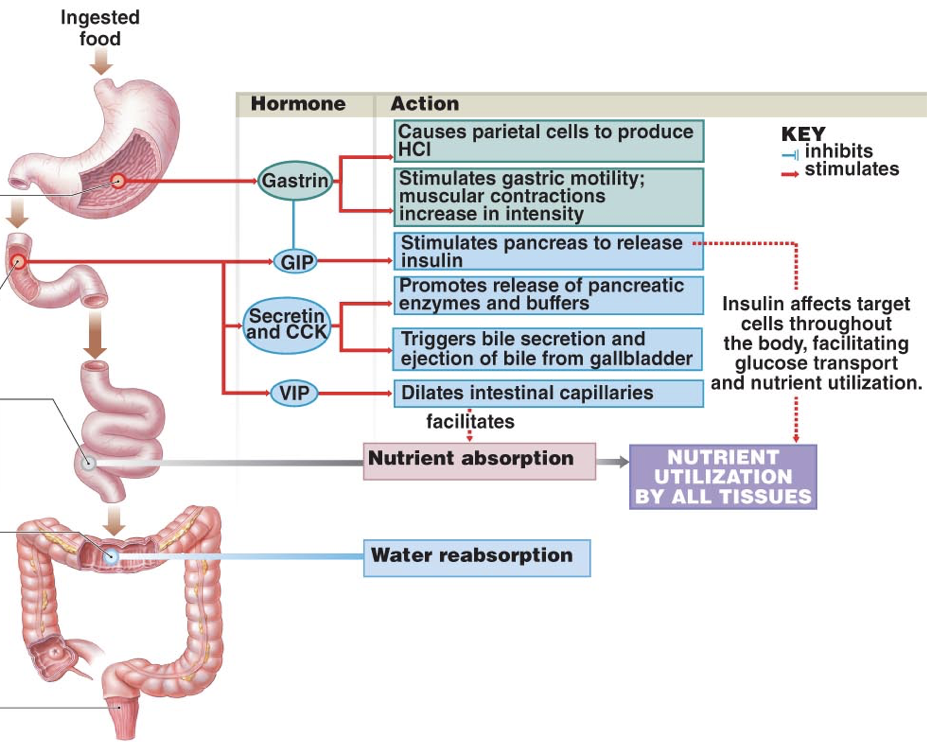 ents causes G cells in the pyloric antrum of the stomach to release gastrin, an endocrine signal with numerous targets. Gastrin is also released by enteroendocrine cells in the duodenum in response to large quantities of incompletely digested proteins and it can also be released by the pancreas.
ents causes G cells in the pyloric antrum of the stomach to release gastrin, an endocrine signal with numerous targets. Gastrin is also released by enteroendocrine cells in the duodenum in response to large quantities of incompletely digested proteins and it can also be released by the pancreas.
Due to autonomic innervation of G cells via the vagus nerve, gastrin release can occur in anticipation of food entering the stomach. For example, smelling food, looking at food (or things that might be food), or even thinking about food, can all stimulate the release of gastrin. In other words, gastrin may be released during the cephalic phase of energy consumption. Gastrin is also released in response to distension of the antrum, circulating epinephrine, and hypercalcemia (via calcium-sensor receptors).
Effects of Gastrin
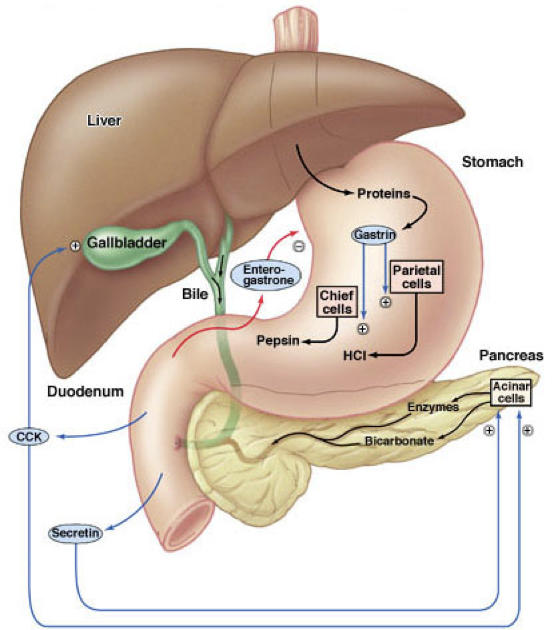
Gastrin has multiple effects on digestive processes within the stomach. Firstly, gastrin enhances the intensity of muscular contractions in the stomach. Gastrin also stimulates the release of pepsinogen by gastric chief cells. Pepsinogen is an inactive proenzyme (a zymogen). Pepsinogen is partially activated when exposed to an acidic environment, after which another partially activated pepsinogen can cleave a peptide from pepsinogen to yield pepsin, an active proteolytic enzyme. The conversion of pepsinogen to pepsin, and the optimal functioning of pepsin, requires an acidic environment. Acidity aids in the digestion of proteins not only by enabling the activation of pepsin, but also by denaturing ingested proteins, which exposes peptide bonds for cleavage by pepsin.
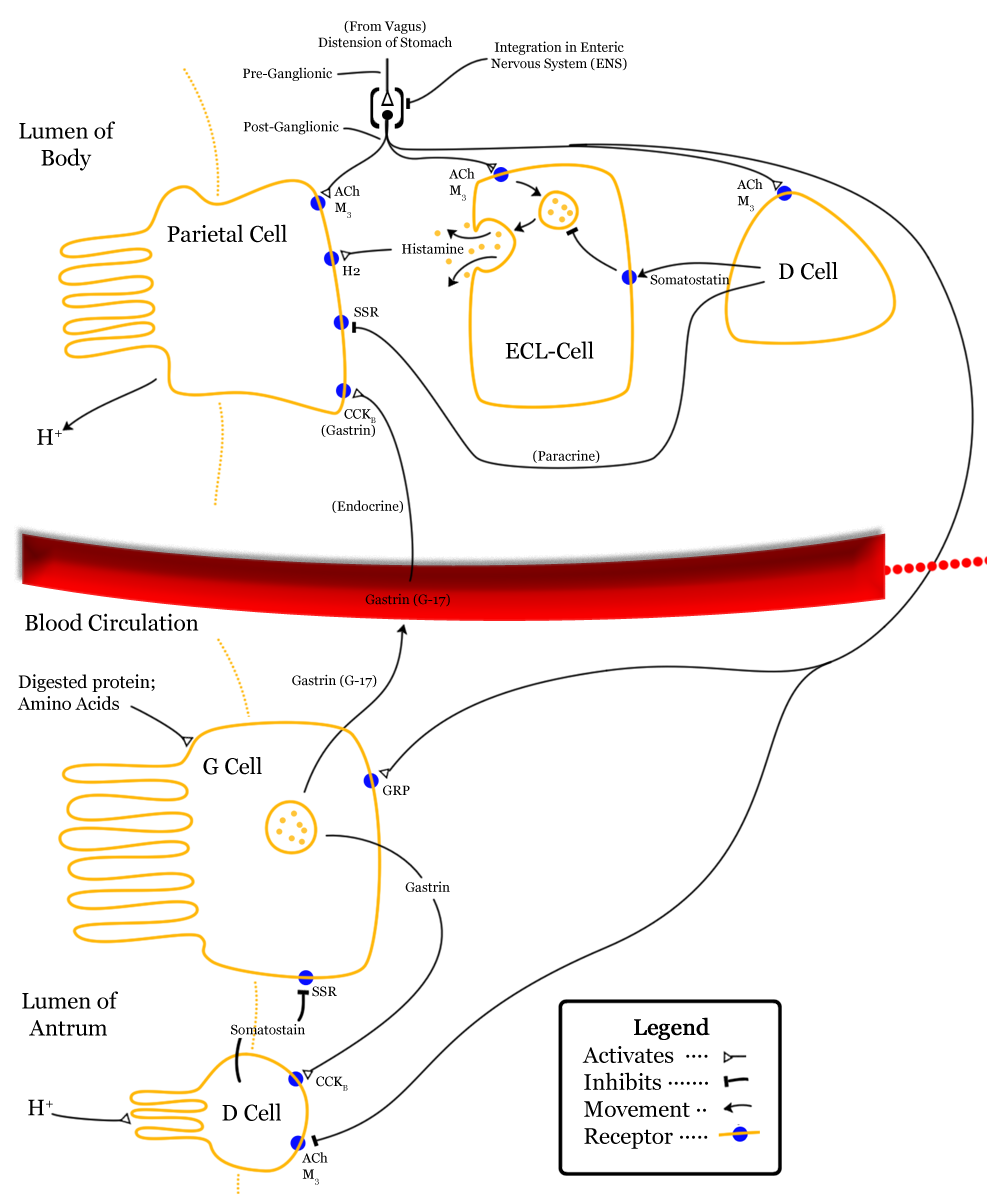
Parietal cells are responsible for creating the acidic environment in the stomach that permits pepsin to cleave proteins with optimal efficiency. To generate the acidic environment in the stomach, parietal cells use hydrogen-potassium-ATPase pumps and chloride-potassium symporters to excrete hydrochloric acid. The hydrogen-potassium-ATPase pumps are unique to parietal cells. They actively exchange extracellular potassium for intracellular hydrogen. This moves hydrogen ions against the strongest ion concentration gradient in the body; hydrogen concentrations in the stomach are about 3-million times greater than in parietal cells. Once potassium is inside the cell, it is paired with chloride and transported out of the cell by chloride-potassium symporters using the concentration gradient of potassium. Collectively, this creates a net efflux of hydrogen and chloride from parietal cells, raising the concentration of hydrochloric acid in the stomach. However, this excretion of hydrochloric acid by parietal cells is regulated by numerous signals, which either drive translocation of the hydrogen-potassium-ATPase pumps to the membrane or promote the removal of hydrogen-potassium-ATPase pumps from the membrane.The role of histamine in the stomach explains how antihistamines alter digestive processes: by blocking histamine-mediated excretion of hydrochloric acid.
Firstly, gastrin both directly and indirectly stimulates translocation of hydrogen-potassium-ATPase pumps to the membranes of parietal cells. Gastrin binding to CCKB receptors can directly stimulate translocation by increasing intracellular calcium concentrations. However, this direct effect of gastrin only has a minor impact on translocation of hydrogen-potassium-ATPase pumps. However, gastrin also stimulates enterochromaffin-like cells (ECL cells), which are neuroendocrine cells that release histamine. Histamine binding to H2 receptors on parietal cells is the most salient stimulus to translocate hydrogen-potassium-ATPase pumps. When histamine binds to G-protein-coupled H2 receptors, there is increased activation of adenylyl cyclase and consequent increases in the concentration of cAMP. Increased cAMP increases the activation of protein kinase A (PKA), which phosphorylates proteins involved in the translocation of hydrogen-potassium-ATPase pumps to the membrane. In addition to releasing histamine in response to gastrin, ECL cells also release histamine in response to activation of M3 receptors by acetylcholine from the vagus nerve. The vagus nerve also innervates parietal cells, where M3 receptor activation promotes hydrogen-potassium-ATPase pump translocation by increasing intracellular calcium concentrations; this effect on translocation is weak in comparison to histamine-mediated translocation. However, gastrin, histamine, and acetylcholine operate synergistically to promote hydrochloric acid excretion. When operating in concert, the effects of gastrin, histamine, and acetylcholine are greater-than-additive.
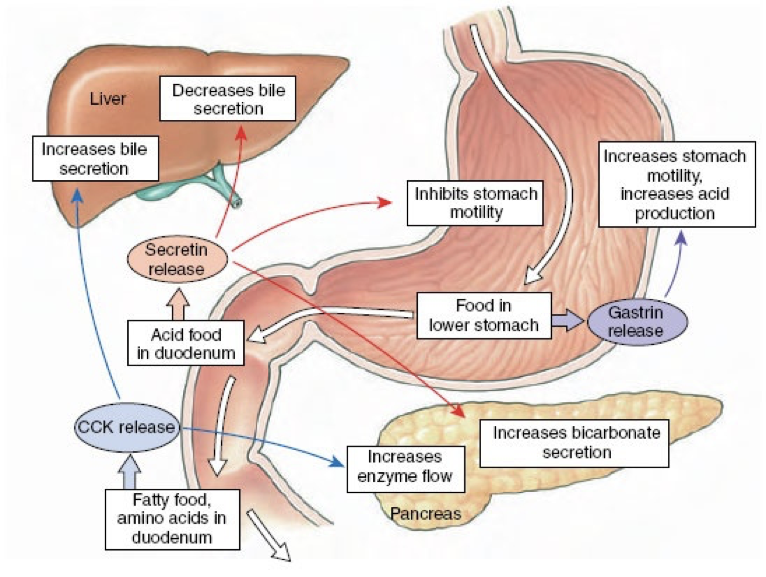
Importantly, gastrin is a signal to the pancreas to release insulin. This is important because the effects of insulin are relatively slow acting compared to the increases in blood sugar as glucose is absorbed. Through gastrin’s stimulation of insulin release, insulin can signal cells to uptake sugar in preparation for an increasing blood sugar, thus dampening the fluctuations in blood sugar levels during and after a meal. However, gastrin’s release is driven largely by the presence of proteins, not sugars, so eating high-protein, low-sugar meals can lead to an unnecessarily large amount of insulin for the amount of sugar, and ultimately lead to hypoglycemia. An easy solution is to eat a sugar-rich desert following a healthy meal.
In addition to its effects on hydrochloric acid excretion (both direct and indirect), pepsinogen excretion, and insulin secretion, gastrin also increases the rate of gastric emptying due to the strength of contractions against the pylorus and by relaxing the pyloric sphincter. Gastrin also promotes excretion of bile by the gallbladder and is involved in the relaxation of the ileocecal valve separating the small intestine from the large intestine.
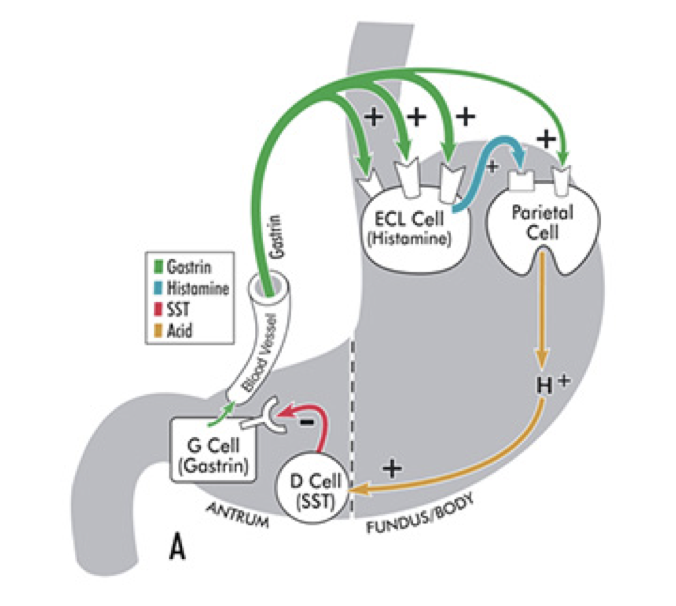
Somatostatin: An Inhibitor of Gastrin
Gastrin release is inhibited by multiple signals, including secretin, gastric inhibitory peptide, vasoactive intestinal peptide, glucagon, and calcitonin. Despite their roles in raising the acidity of the stomach, gastrin release and histamine release are inhibited by high acidity in the stomach. Once the stomach is acidic, there is no longer a need for gastrin and histamine to promote further acidity. However, the acidity does not directly inhibit gastrin and histamine release. Instead, they are inhibited by somatostatin (SST). In addition to being produced by δ cells in pancreatic islets, somatostatin is also produced by δ cells in the pyloric antrum (and to a lesser degree in the duodenum). When exposed to high acidity in the stomach, δ cells release somatostatin into open circulation, at which point somatostatin inhibits the release of gastrin, inhibits the release of histamine, and directly inhibits parietal cells’ excretion of hydrochloric acid; all of these inhibitory influences minimize the excretion of hydrochloric acid into the stomach. Whereas histamine stimulates parietal cells’ excretion of hydrochloric acid through activation of adenylyl cyclase, somatostatin directly opposes this action; somatostatin receptor activation on parietal cells inhibits adenylyl cyclase. This is a form of negative feedback.
In addition to being stimulated by acidity, δ cells in the pyloric antrum are also excited by gastrin itself, leading to a form of autoregulation within the stomach. δ cells also receive cholinergic input from the vagus nerve (also via M3 receptor activation). However, while M3 receptor activation on ECL cells promotes histamine secretion, M3 activation on δ cells inhibits somatostatin secretion.
Other Effects of Somatostatin
Beyond inhibiting the secretion of gastrin and histamine, somatostatin also inhibits the secretion of secretin, CCK, gastric inhibitory peptide, vasoactive intestinal peptide, insulin, and glucagon, while also inhibiting pancreatic excretions. Somatostatin is also important in processes other than digestion. Somatostatin is also released by the hypothalamus into the hypophyseal portal system, which carries somatostatin to the anterior pituitary. At the anterior pituitary, somatostatin inhibits the release of growth hormone, thyroid stimulating hormone, and prolactin. Somatostatin release by the hypothalamus is stimulated by growth hormone, enabling growth hormone to exert negative feedback on its own release through somatostatin secretion.
Release of Secretin
After chyme (partially digested food) passes from the stomach through the pyloric sphincter, it enters the duodenum (the beginning of the small intestine). Chyme entering the duodenum of the small intestine is acidic due to the low pH environment of the stomach. The acid in the chyme stimulates the release of secretin by the duodenum. Secretin was the first hormone identified (first discovered in 1902). Secretin is produced by S cells in the duodenum and to a lesser degree in the jejunum. S cells release secretin when they are exposed to a low pH environment (pH < 4.5). One of secretin’s predominant effects is to raise the pH of the chyme beyond a neutral level to an alkaline level (~7.6-7.8). This allows the enzymes in the small intestine, which require a higher pH, to function optimally.
Effects of Secretin
To raise the pH in the duodenum, secretin inhibits hydrochloric acid excretion by parietal cells, and stimulates excretion of sodium bicarbonate by the pancreas. Secretin inhibits hydrochloric acid excretion by parietal cells directly, but also indirectly inhibits hydrochloric acid excretion by parietal cells by stimulating the release of somatostatin and inhibiting the release of gastrin. Secretin strongly stimulates the excretion of sodium bicarbonate by ductal cells of the pancreas. Furthermore, secretin promotes the excretion of water and bicarbonate from duodenal cells. Collectively, these effects raise the pH of chyme in the duodenum.
Secretin also has effects beyond raising the pH of chyme in the duodenum. Secretin stimulates the production of bile by the liver (which is then stored in the gallbladder). However, low levels of secretin inhibits the excretion of bile by the gallbladder at first (though this inhibitory effect is relatively weak). Then becomes excitatory as levels increase. Secretin also promotes insulin release and serves as a peripheral satiety signal.
In addition to its production in S cells, secretin is also produced by magnocellular neurons in both the paraventricular nucleus of the hypothalamus and in the supraoptic nucleus of the hypothalamus. Secretin in these magnocellular neurons is released from the posterior pituitary in response to increased osmolarity. Hypothalamic secretin also stimulates vasopressin release and mediates the release of vasopressin in response to angiotensin II.
Release of Cholecystokinin
 If the chyme entering the duodenum and jejunum contains fat, the presence of fat triggers the release of cholecystokinin (CCK) by I cells. However, CCK is also released by I cells in response to acetylcholine released by the vagus nerve. CCK is both a central neuropeptide and a gastric peptide hormone. In addition to its release by I cells in the duodenum and jejunum, CCK is also released by enteric nervous system neurons as well as by neurons in the brain.
If the chyme entering the duodenum and jejunum contains fat, the presence of fat triggers the release of cholecystokinin (CCK) by I cells. However, CCK is also released by I cells in response to acetylcholine released by the vagus nerve. CCK is both a central neuropeptide and a gastric peptide hormone. In addition to its release by I cells in the duodenum and jejunum, CCK is also released by enteric nervous system neurons as well as by neurons in the brain.
Effects of Cholecystokinin
CCK is similar in structure to gastrin, and both serve as agonists at the CCKA and CCKB receptors. CCK binding to CCKA receptors on pancreatic acinar cells stimulates their excretion of enzymes and buffers into the pancreatic duct. CCK also stimulates contraction of the gallbladder via CCKA receptor activation, resulting in the ejection of bile (including bile salts) into the bile duct. When there are lipids present in the duodenum to stimulate the release of CCK, CCK overcomes the weak secretin-mediated inhibition of bile excretion. The pancreatic and bile duct converge, becoming the ampulla of Vater (also called the hepatopancreatic duct). The sphincter of Oddi (also called the hepatopancreatic sphincter) controls whether the contents of the ampulla of Vater enter the duodenum. CCK relaxes the sphincter of Oddi, allowing enzymes and buffers (from pancreatic acinar cells) and bile (from the gallbladder) to enter the duodenum. Bile salts from the gallbladder and pancreatic lipase operate in concert to facilitate the digestion and absorption of lipids in the small intestine.
Major pancreatic enzymes include pancreatic α-amylase, pancreatic lipase, nucleases, and proteolytic enzymes. Proteolytic enzymes are not secreted in their active form. Rather, they are secreted as inactive proenzymes (zymogens). This protects the pancreas against the action of its own enzymes. Once the proenzymes reach the duodenal lumen, they become active. These proteolytic enzymes include trypsin (from trypsinogen), chymotrypsin (from chymotrypsinogen), carboxypeptidase A, carboxypeptidase B, and elastase. Together, these proteolytic enzymes break down proteins into a mixture of tripeptides, dipeptides, and amino acid monomers. Dipeptidases already present on the walls of the small intestine contribute to protein digestion.
In addition to stimulating pancreatic enzyme and bile excretion, CCK delays gastric emptying, and stimulates the release of somatostatin by δ cells through interactions with the same receptor bound by gastrin. Also like gastrin, CCK release is inhibited by somatostatin, so stimulation of somatostatin release seems to be an autoregulatory mechanism for CCK release. In addition to being inhibited by somatostatin, CCK release is also inhibited by pancreatic polypeptide.
Despite the distribution of CCK receptors throughout the brain, circulating CCK is unable to permeate the blood-brain barrier, so it is only able to interact with regions of the hypothalamus and brain stem that lack the protective blood-brain barrier. Circulating CCK promotes satiety through its effects on the hypothalamus and brain stem, and also through stimulation of vagus nerve afferents.
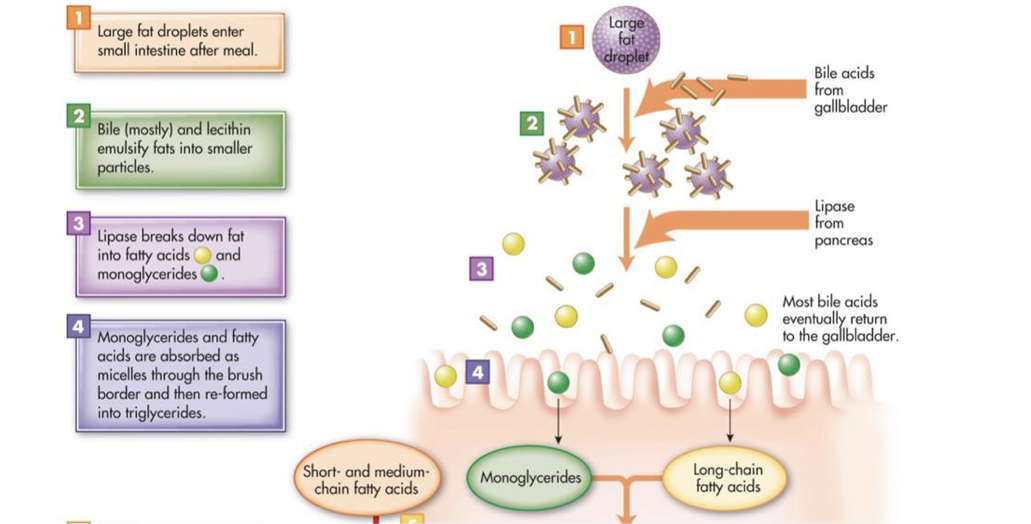
Most consumed fat is triacylglycerol (three fatty acids bound to glycerol). However, triacylglycerol is hydrophobic and is not soluble in the hydrophilic environment of the small intestine. The excretion of bile into the duodenum plays an important role in the lipid digestion by providing bile salts and phospholipids, both of which are amphipathic (i.e., they have both hydrophilic polar regions and hydrophobic nonpolar regions). Intestinal motility breaks down fat globules into small droplets, which are coated in bile salts and phospholipids (this coating prevents the re-association of the small droplets). The nonpolar regions of bile salts and phospholipids face inward towards the nonpolar triacylglycerol molecules, and the polar regions of bile salts and phospholipids face the surrounding hydrophilic environment. In other words, bile salts and phospholipids emulsify fats. The emulsion droplets are the site of triacylglycerol digestion by pancreatic lipase. The water-soluble pancreatic lipase can only operate at the surface of fat globules and emulsion droplets. Since emulsion droplets are much smaller than the fat globules, emulsification dramatically increases the surface area on which pancreatic lipase can operate. Colipase, an amphipathic protein, anchors pancreatic lipase to the surface of emulsion droplets, at which point pancreatic lipase can hydrolyze triacylglycerol molecules into fatty acids and monoglycerides. The resultant fatty acids and monoglycerides (as well as fat-soluble vitamins and cholesterol) associate with bile salts and phospholipids to form micelles, which are about 200-times smaller than emulsion droplets. However, micelles constantly break down and reform. Micelles transport monoglycerides and fatty acids between microvilli to the surface of enterocytes for absorption. While micelles themselves cannot be absorbed, the constant breakdown of micelles releases monoglycerides and fatty acids. These freely-dissolved monoglycerides and fatty acids can diffuse across the membranes of enterocytes. Once in enterocytes, triacylglycerol is synthesized from the absorbed monoglycerides and fatty acids.
Release of Vasoactive Intestinal Peptide
Vasoactive intestinal peptide (VIP) is released by the duodenum (though it is also released by the pancreas as well as the suprachiasmatic nucleus of the hypothalamus). VIP released by the duodenum into circulation facilitates nutrient absorption by the intestines, but VIP also has numerous other effects. To facilitate nutrient absorption, VIP dilates regional capillaries. By dilating capillaries in active areas of the intestinal tract, VIP provides sufficient blood to efficiently uptake absorbed nutrients.
Effects of Vasoactive Intestinal Peptide
VIP also regulates other digestive processes. VIP stimulates the excretion of water into bile and stimulates the excretion of water and bicarbonate by the pancreas. VIP also inhibits gastrin-mediated hydrochloric acid excretion by parietal cells, though VIP stimulates the excretion of pepsinogen by chief cells. Like gastrin and CCK, VIP also stimulates the release of somatostatin by δ cells, which subsequently inhibits the release of VIP. In addition to promoting regional dilation of capillaries, VIP also stimulates cardiac contractility. Despite the increased contractility, VIP tends to lower arterial blood pressure. VIP also relaxes numerous smooth muscles throughout the body, including smooth muscles lining the trachea, smooth muscles of the lower esophageal sphincter, smooth muscles of the gallbladder, and smooth muscles of the stomach. VIP also promotes glycogenolysis, and stimulates prolactin release and growth hormone release by the anterior pituitary.
Finally, central VIP has an important role in the regulation of circadian rhythms. VIP releasing neurons, and neurons expressing VIP receptors, are found in the ventrolateral suprachiasmatic nucleus of the hypothalamus. The suprachiasmatic nucleus of the hypothalamus is the body’s “master clock;” it is the dominant center of circadian control and synchronizes the body’s circadian rhythms to its own. Neurons in the ventrolateral suprachiasmatic nucleus (those containing VIP and/or VIP receptors) receive retinohypothalamic projections, providing these neurons with signals of environmental light conditions. These neurons can then co-release VIP and γ-aminobutyric acid (GABA), which helps synchronize the suprachiasmatic nucleus to the light-dark cycle.
Release of Gastric Inhibitory Peptide and Glucagon-Like Peptide 1
Gastric inhibitory peptide (GIP) is secreted by K cells of the duodenum and jejunum when fats and carbohydrates, especially glucose, enter the small intestine, indicating an impending increase in blood sugar. GIP inhibits gastrin (thereby decreasing hydrochloric acid excretion by parietal cells), delays gastric emptying, stimulates lipid synthesis in adipose tissue, and increases glucose use by skeletal muscles. However, the most important role of GIP is to enhance pancreatic β cells’ glucose-dependent release of insulin. In fact, GIP is also referred to as glucose-dependent insulinotropic polypeptide (still conveniently abbreviated as GIP).
Like GIP, glucagon-like peptide 1 (GLP-1) also enhances glucose-dependent insulin secretion. GLP-1 is produced through post-translational processing of proglucagon in intestinal L cells, which are found in increasing concentrations from the duodenum to the colon (i.e., they are most concentrated in the ileum). Proglucagon is synthesized in both intestinal L cells as well as in pancreatic α cells. However, proglucagon undergoes different post-translational processing in intestinal L cells and pancreatic α cells. In intestinal L cells, proglucagon is cleaved into GLP-1, but in pancreatic α cells, proglucagon is cleaved into glucagon. Both GIP and GLP-1 are members of the glucagon peptide family, and both are degraded by dipeptidyl peptidase 4 (DPP4).
Following food consumption, GLP-1 plasma concentrations rise within minutes. However, GLP-1 released by L cells undergoes local degradation by DPP4. Surviving GLP-1 passes through the hepatic portal to the liver, at which point there is further degradation by hepatic DPP4. As a consequence of the local and hepatic degradation by DPP4, only 10-15% of endogenous GLP-1 reaches systemic circulation. The rapid degradation by DPP4 is largely responsible for the very short 1-2-minute half-life of GLP-1. GIP is also degraded by DPP4, but has a longer, 7-minute half-life. GLP-1 that is not degraded by DPP4 is cleared by the kidneys.
Effects of Gastric Inhibitory Peptide and Glucagon-Like Peptide 1
Once in systemic circulation, GLP-1 is able to interact with GLP-1 receptors (GLP-1R), which are expressed in the pancreas, kidneys, heart, lungs, adipose tissue, smooth muscles, and specific nuclei within the central nervous system. GLP-1R activation on pancreatic β cells is responsible for potentiating glucose-dependent insulin secretion.
The Insulin Effect
GIP and GLP-1 are both incretins and are largely responsible for the incretin effect. If oral glucose consumption (which stimulates release of incretins) and intravenous glucose infusion (which does not stimulate release of incretins) both result in equal blood glucose levels, insulin secretion following oral glucose consumption will exceed insulin secretion following intravenous glucose infusion. The difference in insulin secretion in these two conditions (oral glucose consumption versus intravenous glucose infusion) is due to the effects of incretins on glucose-dependent insulin secretion. In other words, the incretin effect is the enhancement of glucose-dependent insulin secretion that occurs in response to a meal. While the presence of glucose governs insulin release, the quantity of insulin released in response to a given concentration of glucose is largely influenced by incretins. The incretin effect is vital for efficient blood glucose regulation. In fact, the incretin effect is responsible for up to 70% of insulin secretion following oral glucose ingestion. Most of this incretin effect is attributable to GIP and GLP-1. Keep in mind that the incretin-enhanced secretion of insulin remains glucose-dependent. The incretin effect is only present when blood glucose levels are sufficiently high to stimulate insulin secretion. In other words, glucose stimulates the release of insulin, but the quantity and efficacy of insulin release in response to glucose is greater when β cells are stimulated by incretins.
(Figure) Role of GLP-1 and GIP in Regulating Gastric Emptying and Glucose Homeostasis
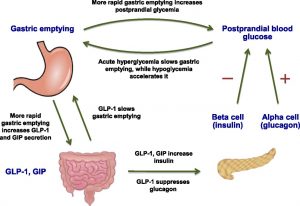
Shortly after consuming food, gastric emptying (when food leaves the stomach and enters the duodenum of the small intestine) occurs, leading to the absorption of glucose into the bloodstream. The rate of gastric emptying is regulated from both directions such that it does not become too high nor too low during food digestion. Under acute hyperglycemia (high blood glucose), gastric emptying is slowed down. Conversely, in the case of hypoglycemia (low blood glucose), gastric emptying accelerates.
Regardless of whether hyperglycemia or hypoglycemia is the case, gastric emptying still occurs and blood glucose levels will rise as a result (although with hyperglycemia, the rise in glucose levels is much slower). As such, a mechanism to help lower the blood glucose levels during gastric emptying is critical for glucose homeostasis. First, gastric emptying increases the signaling for the secretion of two incretins (this process is known as postprandial activation): GLP-1 and GIP. While GLP-1 and GIP both stimulate the release of insulin from the pancreas, GLP-1 also suppresses glucagon activity and slows down gastric emptying (resulting in a negative feedback loop). Since GLP-1 and GIP help lower blood glucose levels significantly, the secretion of these incretins are proportional to the rate of gastric emptying (faster gastric emptying leads to the release of more GLP-1 and GIP). Together, the incretins GLP-1 and GIP contribute to the regulation of gastric emptying and the homeostasis of blood glucose.
Effects of GLP-1 on Pancreatic α and β cells
On β cells, GLP-1R binding activates adenylyl cyclase, yielding a consequent increase in the production of cAMP. Accumulating cAMP activates protein kinase A (PKA), which alters ion channel activity that results in increased intracellular calcium. When levels of intracellular calcium are increased, insulin release in response to glucose influx is potentiated. GLP-1R activation in β cells also promotes insulin transcription, ensuring sufficient availability of insulin for release in response to glucose influx. Furthermore, GLP-1R activation promotes β cell proliferation and regeneration while inhibiting apoptosis. Collectively, these effects increase β cell mass.
In addition to potentiating the glucose-dependent release of insulin, GLP-1 also suppresses glucose-dependent glucagon release by pancreatic α cells. However, this suppression of glucagon secretion is not necessarily achieved through direct inhibitory effects on pancreatic α cells. GLP-1 may have some direct effects on pancreatic α cells, but it is more likely that GLP-1 inhibits glucose-dependent glucagon secretion by increasing secretion of somatostatin from neighboring pancreatic δ cells and insulin from neighboring β cells, both of which inhibit glucose-dependent glucagon secretion by pancreatic α cells.
Effects of GLP-1 on Gastric Glucose Absorption and Gastric Emptying
While the insulinotropic effects of GLP-1 have a major impact on blood sugar reduction by enhancing the storage of glucose present in the bloodstream, GLP-1 also has a significant impact on the rate at which glucose is absorbed into the bloodstream. GLP-1 delays gastric emptying, which slows the rate at which nutrients, including glucose, are absorbed. As a consequence of delayed gastric emptying, postprandial blood glucose rises more gradually, reducing insulin demand. This enables more efficient insulin-mediated glucose storage and minimizes the degree of postprandial hyperglycemia. This delay in gastric emptying is commonly referred to as the ileal brake. GLP-1 also inhibits hydrochloric acid excretion in the stomach.
Some receptors behave differently in response to different rates and durations of stimulation. As an example, the delay in gastric emptying resulting from GLP-1R activation only occurs when GLP-1Rs are intermittently activated (as would occur in response to endogenously-released GLP-1). The delay in gastric emptying does not occur if GLP-1Rs are continuously activated, as would be the case if an individual takes a GLP-1R agonist that remains active in circulation for a long period of time (e.g., more than a few days).
GLP-1R and the Renal Systems
GLP-1R activation also affects the renal systems. In the kidneys, GLP-1R activation decreases the retention of sodium (i.e., GLP-1 promotes natriuresis) by reducing activity of sodium-hydrogen exchanger 3 in proximal convoluted tubules. This natriuretic effect may not be mediated by direct GLP-1R activation in the kidneys. Instead, GLP-1 may modulate the renin-angiotensin-aldosterone signaling pathway, or GLP-1 may modulate neural pathways regulating renal filtration.
GLP-1’s Role in the Brain
In addition to these peripheral effects, circulating GLP-1 is also capable of permeating the blood-brain barrier and stimulating GLP-1Rs in the central nervous system. The GLP-1R is widely expressed in the central nervous system, including in the circumventricular organs, the amygdala, the ventrolateral medulla, the nucleus of the solitary tract, the thalamic paraventricular nucleus, the hippocampus, the cortex, the parabrachial nucleus (a regulatory center for feeding behavior), and the hypothalamus (particularly in hypothalamic regions involved in energy homeostasis). This permits peripheral GLP-1 to In addition to the roles of GLP-1 in regulating energy homeostasis at the level of the hypothalamus, another interesting hypothalamic effect of GLP-1R activation is to promote the secretion of gonadotropin releasing hormone (GnRH) into the hypophyseal portal system, at which point GnRH can stimulate gonadotropin release from the anterior pituitary. This promotion of GnRH secretion is accomplished through an interesting signaling pathway. In GnRH neurons, there is continuous production of 2-arachidonoylglycerol (2-AG, an endocannabinoid) by the enzyme diacylglycerol lipase (DGL) and a tonic retrograde release of 2-AG. The tonic retrograde release of 2-AG inhibits excitatory GABAergic inputs (GABA is usually inhibitory, but GABA is excitatory to GnRH neurons). GLP-1R activation on GnRH neurons increases the production of anandamide (another endocannabinoid). Anandamide in GnRH neurons activates TRPV1 receptors, leading to the inhibition of DGL. This prevents the production and tonic retrograde release of 2-AG. Furthermore, GLP-1R activation also increases the activity of adenylyl cyclase, leading to increased cAMP production, and the consequent activation of protein kinase A (PKA). PKA activates nitric oxide synthase (nNOS), which produces nitric oxide (NO). Nitric oxide is released as a retrograde signal to the excitatory GABAergic inputs, where it promotes GABA release. Thus, GLP-1R activation suppresses 2-AG-mediated inhibition of GABAergic excitation, while also promoting GABAergic excitation through retrograde NO signaling. Both of these effects of GLP-1R activation increase the excitatory GABAergic input to GnRH neurons, resulting in increased excitation of GnRH neurons.
promote satiety at the level of the hypothalamus and brainstem. In addition to the peripheral secretion of GLP-1 as an endocrine hormone, GLP-1 is also produced by neurons of the nucleus of the solitary tract (NTS) and the reticular nucleus of the medulla oblongata, which use GLP-1 as a neurotransmitter. In addition to its central roles in the regulation of energy balance, central GLP-1 also appears to serve neuroprotective and neurogenic roles.
Are Incretins a Viable Treatment option for Type 2 Diabetes Mellitus and Obesity?
Due to their insulinotropic and satiety-promoting effects, incretin signaling pathways have been examined as potential therapeutic targets for the treatment of type 2 diabetes mellitus (T2DM) and obesity. While GIP-mediated enhancement insulin secretion is practically nonexistent in T2DM, the effects of GLP-1 are partially maintained; GLP-1 is still able to reliably enhance glucose-dependent insulin secretion, but with reduced potency. Given that degradation of by DPP4 and renal clearance of GLP-1 are unchanged in T2DM, the reduced potency of GLP-1 signaling in T2DM appears to arise from decreased β cell sensitivity to GLP-1.
Due to its reliable (albeit reduced) incretin effect, GLP-1 signaling has received more attention than GIP as a viable target for the treatment of T2DM and certain related disorders. By potentiating glucose-dependent insulin secretion, impairing glucose-dependent glucagon secretion, and delaying gastric emptying, exogenous administration of GLP-1 can completely normalize blood glucose in T2DM patients. However, GLP-1 itself is not a practical treatment for T2DM and related disorders because of its short half-life.
Due to the significant reduction in circulating GLP-1 attributable to degradation by DPP4, DPP4 inhibitors are a promising therapeutic agent The delay in gastric emptying induced by amylin is most likely achieved through central effects on the dorsal vagal complex of the hindbrain. The dorsal vagal complex contains multiple nuclei and plays a critical role in regulating gastric motility. The dorsal vagal complex includes the area postrema, the nucleus of the solitary tract (NTS), and the dorsal motor nucleus of the vagus nerve (DMV). The area postrema mediates some of the physiological and behavioral effects of amylin, and projects to the NTS. The NTS integrates both neural and endocrine signals (including amylin) and relays the integrated signals to the DMV. The DMB projects preganglionic axons toward the stomach, providing autonomic control over gastric motility. One proposed (though not yet validated) mechanism by which amylin delays gastric emptying is through excitation of the NTS. Amylin may accomplish this excitation of the NTS by interacting with the area postrema to promote the area postrema’s excitation of the NTS (possibly complimenting excitatory glutamatergic vagal input to the NTS) or by directly exciting the NTS. When the NTS is excited by amylin (whether directly or indirectly), the NTS could then modulate the activity of the DMV, resulting in DMV-mediated reductions in gastric motility and delayed gastric emptying. In support of this proposed pathway, gastric emptying is accelerated during hypoglycemia to increase the rate of glucose absorption to raise blood glucose to normal levels. Amylin-activated neurons in the area postrema have been shown to exhibit significant reductions in activity when exposed to low plasma glucose, which, through the same proposed pathway, would yield the increased rate in gastric emptying evident in hypoglycemia. Hypoglycemia may also diminish these neurons’ responsiveness to amylin.
for the treatment of hyperglycemic disorders such as T2DM. GLP-1R agonists (GLP-1RAs) have also received considerable attention.
Peptide YY
In addition to releasing GLP-1, L cells also secrete peptide YY (PYY) in response to nutrients in the small intestine. PYY interacts with neuropeptide Y receptors (NPY receptors). PYY inhibits gastric motility, which, like GLP-1, slows the rate of nutrient absorption. PYY also increases the absorption of water and electrolytes in the large intestine, and promotes satiety. In addition to its peripheral release by L cells, PYY is also used as a neurotransmitter by neurons in the gigantocellular reticular nucleus of the medulla.
Secretion and Effects of Amylin
By enhancing the glucose-dependent secretion of insulin, GLP-1 and GIP also enhance the secretion of amylin by pancreatic β cells. Amylin is cosecreted with insulin from pancreatic β cells in a ratio of roughly 1 amylin per 100 insulin. In addition to the pancreas (the primary peripheral site of amylin production), peripheral amylin is also expressed in the lung, gastrointestinal tract (including the stomach and pylorus), and dorsal root ganglia. The effects of amylin are complimentary to the effects of insulin. While there is not necessarily a direct functional interaction, amylin and insulin both have blood sugar-reducing effects. The peripheral effects of amylin and GLP-1 largely overlap. Like GLP-1, amylin delays gastric emptying, which slows the rate of glucose absorption and thereby dampens postprandial rises in serum glucose. Also like GLP-1, amylin inhibits glucagon secretion. Peripheral amylin has a half-life of 13 minutes, and is primarily deactivated by renal clearance. This likely involves degradation of amylin by insulin-degradinPramlintide is an amylin agonist approved for the treatment of T1DM and T2DM. Pramlintide is co-administered with insulin analogs and contributes to suppression of postprandial blood glucose by delaying gastric emptying, inhibiting glucagon secretion, and reducing food consumption. Like amylin, pramlintide is primarily deactivated by renal clearance, which also likely involves degradation by insulin-degrading enzyme. In obesity, satiety signals often have a diminished capacity to elicit their anorexigenic effects due to decreased receptor sensitivity. This is particularly true in the case of the adipose tissue-derived satiety signal leptin. In obesity, leptin insensitivity (also called leptin resistance) is profound. However, amylin receptor agonists remain potent appetite-suppressing agents in obese individuals but require continuous administration to effectively treat obesity. Furthermore, in some cases, amylin enhances the anorexigenic effects of other satiety signals (e.g., CCK, PYY, insulin, leptin) when co-administered.
g enzyme, which is highly expressed in the kidneys.
Peripheral amylin can cross the blood-brain barrier to stimulate amylin receptors throughout the brain, especially regions involved in the control of feeding behaviors (and other motivated behaviors). These regions include the mesolimbic reward system (in which amylin interacts with the ventral tegmental area and nucleus accumbens), the hypothalamus, caudal brainstem nuclei involved in feeding behavior, the lateral parabrachial nucleus, and especially the area postrema. Amylin is a satiety signal and likely exerts salient anorexigenic effects through interactions with the area postrema. However, amylin receptor activation in the hypothalamus also plays critical roles in amylin’s effects on energy homeostasis. Like certain other anorexigenic hormones, amylin also increases energy expenditure. Beyond its effects on energy homeostasis, amylin also seems to exert cognitive enhancing effects, but whether this is achieved through direct interactions in the central nervous system is unclear.
In addition to peripheral secretion of amylin, amylin is also used as a neurotransmitter by the lateral parabrachial nucleus, the preoptic area of the hypothalamus, the arcuate nucleus of the hypothalamus, and the lateral hypothalamus (which also receives amylinergic input from the lateral parabrachial nucleus).
Due to the co-secretion of amylin with insulin, the loss of β cells in diabetes type I also entails a loss of amylin. Among other things, this causes food to move through the intestines more rapidly and removes the anorexigenic effects of amylin. The lack of amylin has a significant impact on type I diabetics. This also explains why treatment with just insulin fails to resolve all complications, even when the provision of supplementary insulin is tightly regulated.
In T1DM, amylin levels are extremely low. In T2DM, particularly advanced T2DM, amylin levels are also low. These low levels of amylin in T2DM are associated with the reductions in insulin, with which amylin is co-secreted. As a consequence, individuals with diabetes have an accelerated rate of gastric emptying, which leads to more rapid glucose absorption. Without insulin (in T1DM), or without efficient insulin signaling (in T2DM), diabetics are subject to postprandial hyperglycemia. The rapid rate of glucose absorption that occurs in the absence of amylin can further exacerbate this postprandial hyperglycemia.
Due to the delayed rate of gastric emptying in response to amylin signaling (which minimizes postprandial hyperglycemia) and the metabolic effects of amylin signaling pathways, amylin and amylin agonists are viable treatment adjuncts for diabetes and may also aid in the treatment of obesity due to their body weight-reducing effects.
Orexigenic and Anorexigenic Signaling
Leptin: Appetite Suppression and Energy Expenditure
Leptin is released from adipocytes in proportion to their triglyceride reserves such that higher levels of leptin indicate high amounts of energy available. This is a relatively slow, long-term signal of energy availability. Leptin acts directly on neurons of the hypothalamus to suppress appetite and enhance energy expenditure. In contrast, the absence of leptin is a signal that energy availability is depleted, and that consumption is required to replenish reserves. The lack of leptin contributes to appetite and feeding behavior while also lowering the rate at which energy is utilized. Leptin-signaling-deficient humans crave food and have a slower metabolism, which can precipitate obesity. This can be due to mutations in the gene for leptin, but can also be due to a decreased sensitivity to leptin stemming from an under-expression of hypothalamic leptin receptors, insensitive leptin receptors, an inability for leptin to permeate the blood brain barrier, etc.
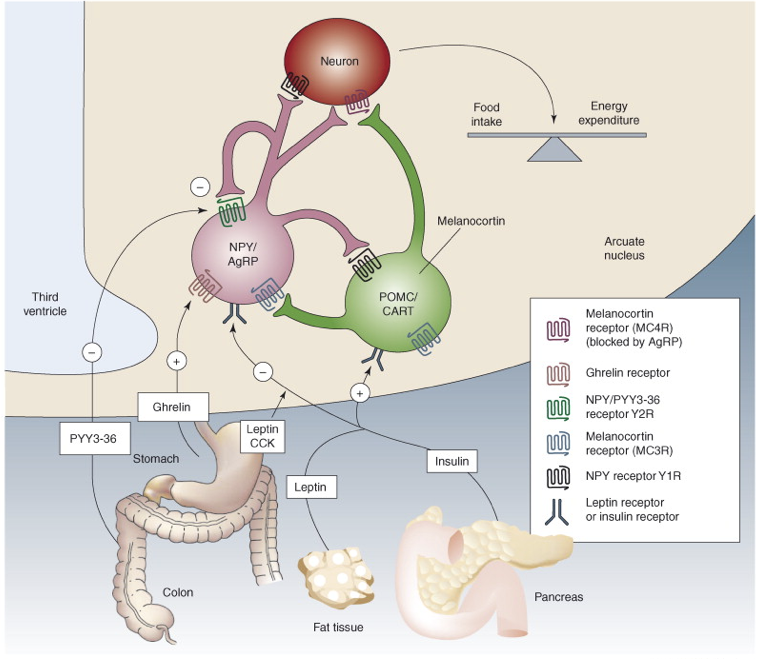
Leptin stimulates POMC neurons of the arcuate nucleus of the hypothalamus, which respond by producing the anorexigenic (appetite-suppressing) peptide transmitters α-melanocyte-stimulating hormone (αMSH) and cocaine- and amphetamine-regulated transcript (CART). αMSH and CART stimulate the paraventricular nucleus (PVN) of the hypothalamus to release of thyrotropin-releasing hormone (TRH) and corticotropin-releasing hormone (CRH), thereby activating the HPT and HPA axes, respectively. The net effect of this pathway is to increase energy expenditure and to increase metabolism. The PVN also projects to preganglionic neurons of the brain stem and spinal cord, including the intermediolateral grey matter, all of which contributes to an increase in sympathetic tone, further increasing metabolism. Lastly, the arcuate αMSH/CART neurons project to the lateral hypothalamus (LH) to inhibit its orexigenic influences on feeding behavior. Thus, the presence of leptin will have an anorexigenic effect, whereas the absence of leptin will prevent activation of these anorexigenic signals, thereby yielding an orexigenic impact.
The roles of leptin in the development of anorexia and obesity are still controversial. Excessive leptin signaling can suppress appetite even when eating is necessary. This is thought to be associated with anorexia, possibly due to hypersensitivity of leptin receptors, which results in a false perception of excessive fat stores. Conversely, a compromised leptin signal seems to increase susceptibility to obesity. Impaired leptin signaling could be the result of a lack of leptin receptors, a failure to produce leptin, a failure to release leptin, etc. A predominant characteristic consequence of obesity is the development of leptin insensitivity (also called leptin resistance) in which leptin receptors no longer bind leptin efficiently. This is comparable to insulin insensitivity (also called insulin resistance) in T2DM.
In contrast, there are also orexigenic neurons in the arcuate nucleus that are inhibited by the presence of leptin, and excited in the absence of leptin. Neuropeptide Y (NPY) and Agouti-related peptide (AgRP) are both produced in some arcuate neurons. These neurons inhibit TRH and CRH neurons of the PVN, and enhance parasympathetic tone through connections in the LH. NPY released to the PVN also results in increased insulin secretion, decreased breakdown of triglycerides in adipose tissue, and decreased body temperature. The effects of these orexigenic NPY/AgRP arcuate neurons are almost directly opposite to those of the anorexigenic effects of signaling by αMSH/CART neurons in the arcuate nucleus. In fact, AgRP is actually an antagonist at inhibitory αMSH receptors (MC4R) found on neurons in the LH, where AgRP blocks the inhibitory effects of αMSH, thereby preventing the appetite-suppressing effects of αMSH.
The LH projects throughout the central nervous system and releases appetite-promoting signals. Some LH neurons receiving arcuate innervation produce the orexigenic signal melanin-concentrating hormone (MCH). OtherAmylin interacts cooperatively with leptin. The anorexigenic effects of lepin when paired with amylin are greater than the sum of leptin and amylin’s independent anorexigenic effects. In other words, there is a greater-than-additive effect on satiety when leptin and amylin signaling pathways are simultaneously activated. The ventromedial nucleus of the hypothalamus (VMH) is one site of amylin-leptin interactions. The administration of amylin enhances leptin binding in the VMH, and both leptin and amylin receptor activation initiate some of the same signaling cascades (e.g., STAT3, Akt, and ERK). The STAT3 pathway seems to be particularly important in the interaction between leptin and amylin. Amylin-enhanced leptin signaling in the VMH requires amylin’s induction of interleukin 6 (IL-6). Notably, even following the development of leptin insensitivity (e.g., in obese individuals), amylin administration can restore leptin sensitivity.
LH neurons produce another orexigenic signal, called orexin (also called hypocretin), which is also involved in the regulation of sleep by promoting wakefulness. Both orexin and MCH levels are inversely related to leptin levels, largely mediated by leptin-responsive arcuate neurons that innervate the LH.
MCH and orexin neurons in the LH project throughout the brain and brainstem, where they innervate regions involved in the generation of motivated behavior, including the cerebral cortex, locus coeruleus, thalamus, reticular formation, periaqueductal gray, and numerous preganglionic ANS neurons. These two orexigenic signals from the LH serve complementary roles, with orexin contributing to initiation of feeding, and MCH sustaining feeding behavior once it is initiated.
Ghrelin: Stimulation of Appetite and Food Consumption
Numerous other signals are involved in regulating food drive. Ghrelin, released as a hormonal signal from the stomach and parts of the duodenum during periods of fasting, stimulates appetite and food consumption by activating arcuate NPY/AgRP neurons. Ghrelin is a relatively short-term cue to the hypothalamus to eat. Ghrelin also exerts orexigenic effects through inhibition of vagus nerve afferents projecting to the nucleus of the solitary tract, which would otherwise signal satiety. Ghrelin is also released within the brain as a neurotransmitter. Since ghrelin drives appetite, its signaling pathway may be a treatment target for certain eating disorders. For example, stomach stapling removes the source of ghrelin. Interestingly, obese people do not have an excessive amount of ghrelin, but they may have modified receptors with higher affinities for ghrelin. This would be a neurological receptor. In contrast, low levels of ghrelin are associated with anorexia. If the problem is the receptors (e.g., low affinity), though, no amount of ghrelin or ghrelin agonists will have a therapeutic effect. In addition to its effects on feeding behavior, ghrelin also stimulates the release of growth hormones.
PYY, released by intestinal L cells, binds to Y2 receptors (Y2Rs) on NPY/AgRP neurons in the arcuate nucleus. Y2Rs are inhibitory autoreceptors agonized by NPY. PYY also acts as an agonist at these Y2Rs on NPY/AgRP neurons, inhibiting their ability to release orexigenic signals, yielding an anorexigenic effect. Unlike some other anorexigenic signals, there is no PYY resistance in obesity, but there are reduced levels of PYY. As a result of the intact functional receptors, exogenous PYY administration may be a viable treatment for obesity.
Anorexigenic Signaling via the Peripheral Nervous System
In addition to hormonal anorexigenic signals, anorexigenic signaling can be conveyed by the peripheral nervous system. During periods of gastric distention following consumption, stretch receptors on the stomach project via the vagus nerve to the nucleus of the solitary tract in the medulla, signaling satiety. This affects the gustatory nucleus (a subdivision of the nucleus of the solitary tract), and also alters the functioning of the autonomic nervous system. CCK is released by intestinal I cells in response to the presence of food, particularly fatty food, in the duodenum. Among its other roles in stimulating digestive hormone and enzyme secretion/excretion, CCK has an inhibitory influence on both meal frequency and size by stimulating vagus nerve afferents projecting to the nucleus of the solitary tract (this effect of CCK directly opposes the effects of ghrelin). Furthermore, CCK interacts with orexin-releasing neurons, which control appetite and wakefulness. These interactions also endow CCK with indirect influence on sleep regulation.
In anticipation of food, vagus nerve efferents stimulate β cell secretion of insulin. This causes a slight decrease in blood glucose levels that activates arcuate NPY/AgRP neurons (among other effects), driving feeding behavior. During feeding, insulin secretion increases further as sugar is absorbed and blood glucose rises. However, insulin secretion is also enhanced by gastric hormones, such as GLP-1 and GIP. While the decreased blood glucose alters the activity of orexigenic NPY/AgRP neurons in the arcuate nucleus and some glucose-sensitive LH neurons, the rising levels of insulin secreted during food consumption has effects on the hypothalamus reminiscent of leptin; it stimulates the ventromedial hypothalamus (VMH), as well as anorexigenic neurons of the arcuate nucleus (POMC neurons), while inhibiting orexigenic arcuate neurons (NPY/AgRP neurons). In fact, insulin’s effects on the hypothalamus tend to have a more salient impact on satiety than leptin.
Lesions to the VMH induce increased neural excitation of insulin secretion, and a general disruption of the balance between the sympathetic nervous system and parasympathetic nervous system. In animals with VMH lesions, the liver and adipose tissue fail to release energy stores during periods of fasting. Without the release of energy stores, animals are forced to eat more frequently to maintain sufficient levels of energy and nutrient availability in the bloodstream. Furthermore, such animals are unusually responsive to the positive (hedonic) aspects of food, and may even seek and consume foods that would otherwise be perceived as nauseating. Overall, VMH-lesioned animals rapidly become obese. However, if the connections to the pancreas are also lesioned, the increase in insulin is resolved, and obesity does not develop.
Conclusion
GLP-1, GIP, insulin, amylin, CCK, and PYY all act as short-term satiety signals to the hypothalamus, and with more pronounced effects on satiety than the effects of leptin. These hormones also act relatively quickly compared to leptin.
To further complicate the generation of feeding behaviors, it appears that different processes underlie the generation of nutritive feeding to maintain energy balance and the generation of hedonic feeding (i.e., eating for pleasure). Destruction of dopaminergic fibers in the LH prevents food seeking behavior without decreasing the pleasure derived from eating. In contrast, stimulation of these dopaminergic fibers tends to promote food-seeking behaviors, though also without altering the pleasure derived from eating. This demonstrates how the palatability of food is attributable to a different circuit than those underlying hunger and food drive.
Other neurotransmitter systems are also involved. Serotonin levels rise in anticipation of food; spike while eating (especially while eating carbohydrate-rich foods), and then fall as food is absorbed. In other words, serotonin generally promotes satiety. This is further supported by studies demonstrating that drugs that enhance serotonergic signaling tend to be anorexigenic. In addition to its proposed role in mood disorders, impaired serotonergic signaling is implicated in hypophagic eating disorders, particularly bulimia nervosa.
These orexigenic and anorexigenic neurotransmitters and hormones have different effects at different synapses and tissues. For example, while it may be a potent factor in feeding, NPY can also suppress ovulation and inhibit sexual behavior.
Feedback/Errata