
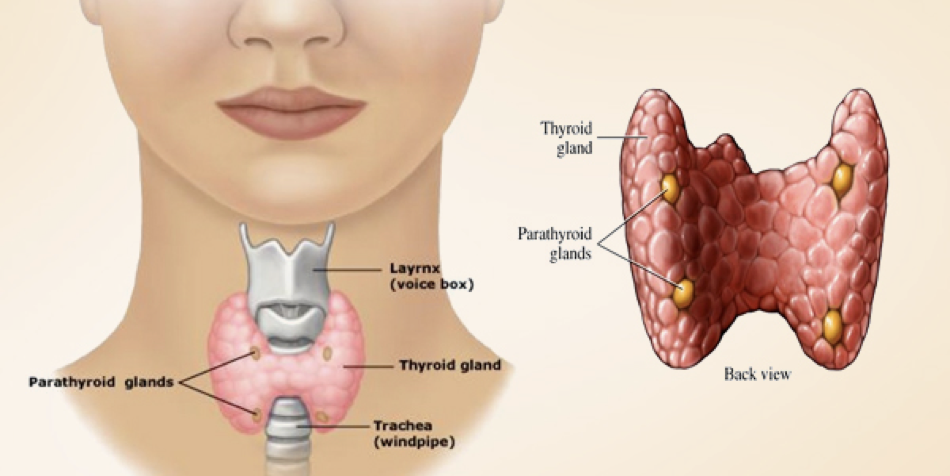
2 Thyroid, Parathyroid, and Calcium Regulation
Calcium Regulation
In addition to its role in metabolic regulation, the thyroid also plays a major role in regulating serum calcium concentration. Calcium is necessary for every cell in the body. The vesicular release of hormones and other signal molecules (e.g., neurotransmitters) relies on calcium; skeletal, cardiac, and smooth muscles all depend on calcium to initiate contractions; bones and teeth use calcium as a fundamental building block; the process by which lipids are liberated from adipose tissue requires calcium; calcium is also necessary for the process of hemostasis, where it is serves essential roles in thrombopoiesis (formation of a blood clot). The vital roles of calcium in these processes (and many more) expound the importance of calcium homeostasis.
The Parathyroid and Calcium Regulation
In addition to the thyroid, the parathyroid is involved in calcium regulation. The parathyroid is composed of four distinct lobes on the dorsal thyroid. In response to low calcium, chief cells in these lobes secrete the parathyroid’s primary hormone, creatively called parathyroid hormone (PTH). PTH induces a rise in serum calcium levels through its interactions with surface receptors on the kidneys that enhance calcium retention, and through a pathway that enhances the release of calcium from bones. PTH also signals the kidneys to convert 25-hydroxy D3 [25(OH)D] to 1,25-dihydroxycholecalciferol [1,25-(OH)2D3, also called calcitriol]. Calcitriol is the active form of vitamin D3, and increases calcium absorption from the intestinal tract while also exerting negative feedback on PTH secretion (we’ll return to calcitriol shortly).
End WG5
The binding of calcium to extracellular calcium sensor receptors (CaSR) on the surface of chief cells governs the release of PTH; low extracellular calcium stimulates the release of PTH, and high extracellular calcium inhibits the release of PTH.
When calcium is in sufficient concentration, it binds to a G-protein-coupled calcium sensor receptor, which has multiple calcium binding sites. When bound by calcium, the receptor activates multiple G-proteins. Some of these G-proteins inhibit adenylyl cyclase, which lowers the concentration of cAMP; other G-proteins initiate MAPK signaling pathways; and other G-proteins activate an IP3 signaling pathway. We’ll focus on the IP3 signaling pathway.
When bound by calcium, the calcium sensor receptor activates phospholipase C (PLC). PLC cleaves PIP2 into DAG and IP3. IP3 binds to receptors on the endoplasmicEnd WG6
reticulum, enabling the release of sequestered calcium. In other words, high external calcium leads to high internal calcium in chief cells. Normally, a rise in internal calcium (e.g., through an IP3 pathway) drives hormone secretion. However, in parathyroid chief cells, the high internal calcium actually inhibits the release of PTH.
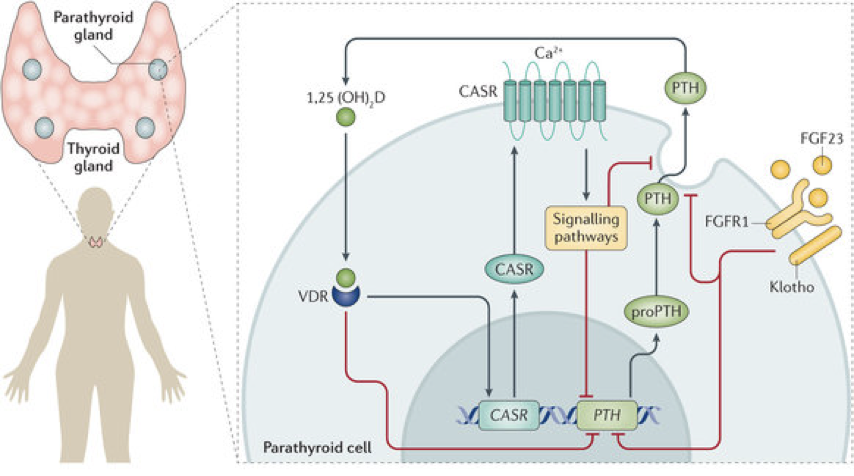 High internal calcium in chief cells enables phosphorylation of phospholipase A2 (PLA2). Phosphorylated PLA2 generates arachidonic acid metabolites, which inhibit PTH secretion. Arachidonic acid metabolites also increase the expression of nuclear calcitriol receptors (vitamin D receptors; VDR) in parathyroid chief cells. Binding of calcitriol to VDR in chief cells both inhibits PTH synthesis (via inhibition of preproPTH synthesis) and sensitizes chief cells to the presence of calcium by increasing calcium sensor receptors’ affinity for calcium (i.e., following VDR activation, the same amount of extracellular calcium will result in greater inhibition of PTH secretion). This is true negative feedback because calcitriol changes chief cells’ responses to the same stimulus.
High internal calcium in chief cells enables phosphorylation of phospholipase A2 (PLA2). Phosphorylated PLA2 generates arachidonic acid metabolites, which inhibit PTH secretion. Arachidonic acid metabolites also increase the expression of nuclear calcitriol receptors (vitamin D receptors; VDR) in parathyroid chief cells. Binding of calcitriol to VDR in chief cells both inhibits PTH synthesis (via inhibition of preproPTH synthesis) and sensitizes chief cells to the presence of calcium by increasing calcium sensor receptors’ affinity for calcium (i.e., following VDR activation, the same amount of extracellular calcium will result in greater inhibition of PTH secretion). This is true negative feedback because calcitriol changes chief cells’ responses to the same stimulus.
Normally, a rise in internal calcium drives hormoneEnd WG7
secretion, but in parathyroid chief cells, the high internal calcium actually inhibits the release of PTH. The signal for PTH secretion is the release of inhibition that results from calcium levels being too low to stimulate the receptor, and consequent low internal calcium.
The importance of properly functioning calcium sensor receptors is exemplified by two calcium regulatory disorders resulting from hyper- or hyposensitive receptors. In familial hypocalciuric hypercalcemia, a hyposensitive calcium sensor receptor requires higher levels of serum calcium to activate the IP3 signaling pathway in chief cells. As a result, the set point for calcium-dependent suppression of PTH secretion rises, leading to chronically elevated levels of serum calcium. In contrast, autosomal dominant hypocalcemia results from expression of a hypersensitive calcium sensor receptor resulting in a lower calcium set point; even low levels of calcium are able to activate the receptor and suppress PTH release. This leads to hypocalcemia.
Magnesium also has an important role in the release pathway of PTH. Magnesium and calcium are both divalent cations, and generally have antagonistic activity. Magnesium is also capable of binding to the calcium receptor on chief cells, though with a much lower binding affinity. Nevertheless, high levels of magnesium can outcompete low concentrations of calcium. This is cause for concern in the event of hypocalcemia. Normally, the low concentrations of calcium would fail to inhibit PTH release, leading to PTH release and a subsequent rise in serum calcium. However, if magnesium levels remain sufficiently high during a period of hypocalcemia, magnesium binding to calcium receptors continues to inhibit the release of PTH. This means that high levels of magnesium have the potential to induce and exacerbate hypocalcemia. In contrast, moderately low levels of magnesium potentiate the effects of PTH on the kidneys. However, very low magnesium levels actually impair PTH secretion. When magnesium levels are too low to bind to the calcium receptor, it appears that the receptor loses functionality and chronically signals the presence of calcium, leading to inhibition of PTH release (i.e., some amount of magnesium is required for PTH secretion). Magnesium is also needed for the production of ATP, and ATP is needed for PTH production. As such, low magnesium may also prevent the synthesis, not just the release, of PTH. Too little magnesium or too much magnesium can lead to hypoparathyroidism, but somewhat low magnesium and very low calcium will drive PTH release most strongly. High levels of calcium, regardless of magnesium, will inhibit the release of PTH.
The Thyroid and Calcium Regulation
Parafollicular cells (also called C-cells) reside in parts of the thyroid surrounding the parathyroid and resemble thyroxin-producing follicle cells. However, they are neuroendocrine cells. When C-cells are activated by high calcium, using the same type of calcium sensor receptor as parathyroid chief cells, downstream pathways promote the secretion of calcitonin, which lowers serum calcium concentrations. In both chief cells and C-cells, activation of the calcium sensor receptor by high extracellular calcium concentrations leads to an increase in IP3 production and a decrease in cAMP production. However, downstream pathways diverge. While calcium sensor receptor activation on parathyroid chief cells ultimately inhibits PTH secretion, calcium sensor receptor activation on parafollicular C-cells actually promotes calcitonin secretion. Like most hormone-releasing cells, high calcium in C-cells is the impetus for secretion. This demonstrates that even the same receptor can have different effects depending on the presence of different intracellular signaling cascades.
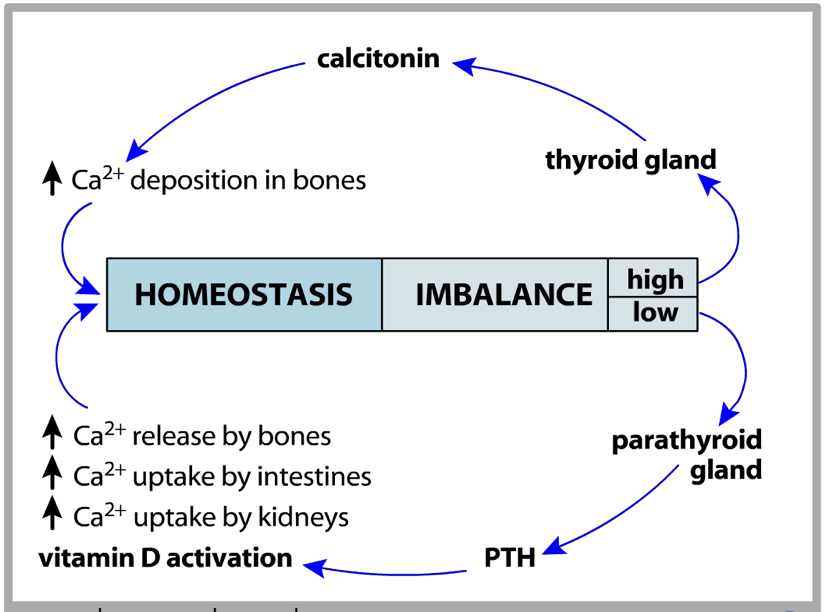
The thyroid and parathyroid are antagonistic in their regulation of calcium. While the parathyroid is responsible for raising serum calcium through PTH secretion, the thyroid is responsible for lowering serum calcium through calcitonin secretion. The thyroid and parathyroid both exert their effects on calcium levels by altering the way in which calcium is stored in bones and the way in which the kidneys reabsorb calcium.
Bones and Calcium Regulation
One of the predominant mechanisms by which the thyroid and parathyroid regulate calcium is by altering the way in which calcium is deposited in bones. Bones are living structures that are constantly growing, retracting, and adapting to physical activity. Some parts of bones are dynamic and change throughout life, while others stop growing at a certain stage of life and remain static from then on. Bones are comprised of cells, cartilage, and mineralized structures of calcium- and phosphate-based apatite. 67% of bones are inorganic, containing calcium and phosphates. The remaining 33% of bones are made up of proteins, primarily collagen. There is very little solid material in bones. In fact, they are remarkably rubbery, and will bend before they break. However, as people age, their bones lose their elasticity. Certain disorders, such as Ehlers-Danlos syndrome, result from a lack of collagen. Among other symptoms, patients with Ehlers-Danlos syndrome may exhibit spinal curvature, loose joints, poor muscle tone, hearing loss, and hypermobility. This is due to a loss of collagen type 3 or 5. In contrast, loss of collagen type 1 can result in fragile long bones, muscle weakness, and blue-colored sclera; this is a condition called osteogenesis imperfecta, or more frequently, brittle bone disease. One of the most significant roles of bones is to store calcium.
At the end of long bones is a region called the growth plate (also called the epiphyseal plate), where cartilage is produced by chondrocytes. The growth plate also gives rise to cells called osteoblasts, which in turn, give rise to osteocytes or osteoclasts.
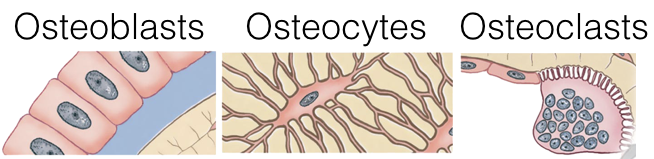
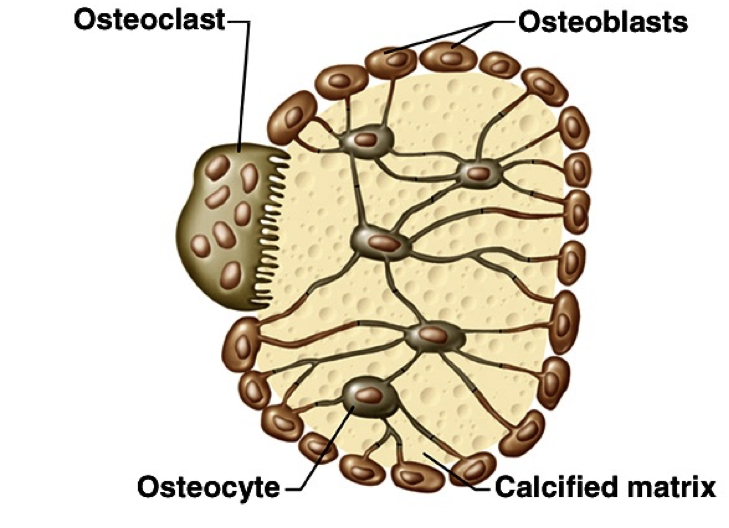 Osteoblasts lay down bone material until they are surrounded by bone, at which point they become osteocytes. Osteoclasts remove and wear-down bone material when necessary; they are involved in remodeling bones. In order to accomplish this, osteoclasts catalyze bone catabolism (degradation) by secreting hydrogen ions into a confined area with active transport pumps to raise the acidity and dissolve bone. The ruffled border on osteoclasts increases surface area to help make this process feasible.
Osteoblasts lay down bone material until they are surrounded by bone, at which point they become osteocytes. Osteoclasts remove and wear-down bone material when necessary; they are involved in remodeling bones. In order to accomplish this, osteoclasts catalyze bone catabolism (degradation) by secreting hydrogen ions into a confined area with active transport pumps to raise the acidity and dissolve bone. The ruffled border on osteoclasts increases surface area to help make this process feasible.
As osteoclasts eat away at bone, osteoblasts rebuild it, causing the constant changes in bones throughout the lifespan. For bones to maintain a specific concentration of calcium, there must be a balance between the activity of osteoblasts and osteoclasts. If osteoblasts are more active, there will be a net increase in bone (loss of blood calcium), whereas if osteoclasts are more active, there will be a net loss of bone (increase in blood calcium).
Bone Growth Disorders
Before puberty, bone growth is regulated by a series of growth hormones. Two of which are human growth hormone and fibroblast growth factor. Human growth hormone (HGH) controls the length of bones. The gene for HGH was the first gene to be cloned. Fibroblast growth factor (FGF) controls the maturity of bones.
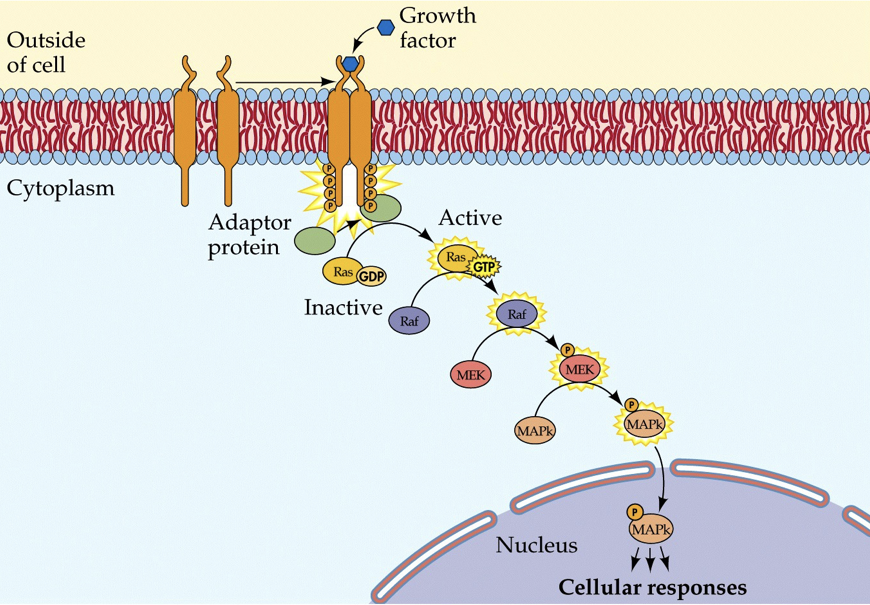 When HGH is present on the outside of the membrane (and when permitted by thyroid hormones), it will cause two transmembrane monomers to dimerize (come together). Both proteins have a receptor for HGH, and when they each bind to a single HGH molecule, they bind to each other (this is different than insulin receptors, which require two insulin molecules to activate a single receptor complex). If the monomers are together for a long enough period, they will phosphorylate each other. This will initiate an enzymatic pathway that signals growth.
When HGH is present on the outside of the membrane (and when permitted by thyroid hormones), it will cause two transmembrane monomers to dimerize (come together). Both proteins have a receptor for HGH, and when they each bind to a single HGH molecule, they bind to each other (this is different than insulin receptors, which require two insulin molecules to activate a single receptor complex). If the monomers are together for a long enough period, they will phosphorylate each other. This will initiate an enzymatic pathway that signals growth.
Human growth hormone (with the permissive action of thyroid hormones) stimulates osteoblast activity, leading to an astonishing rate of calcium deposition prior to adolescence. Acromegaly (a form of gigantism) is the result of an overabundance of HGH, either due to a lack of regulation by the pituitary, or due to a tumor (it is most often caused by a pituitary adenoma). High HGH causes all organs to enlarge and can even result in a goiter due to an enlarged thyroid. This also increases thyroxin concentrations sufficiently to permit the functioning of higher levels of HGH. However, acromegaly is not the only form of gigantism. While some forms involve all bones being bigger (as well as all other structures within the body), other forms only alter the growth of long bones.
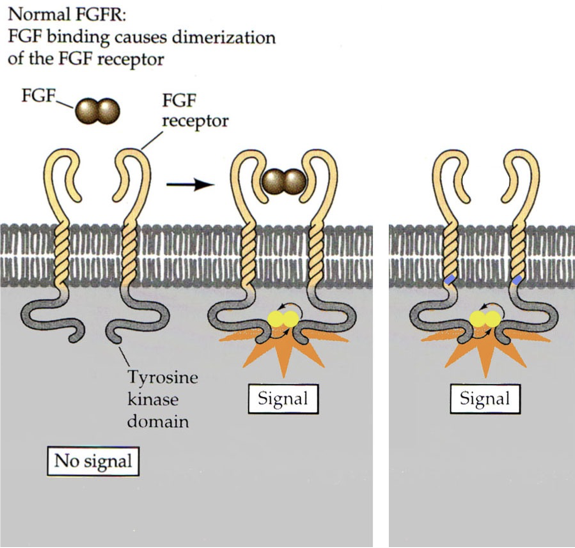 FGF signals that the body is finished growing by instructing chondrocytes to differentiate into bone. Usually, the signal is not released until after puberty. However, a single missense mutation on the dominant allele for FGF receptor 3 can cause the 380th amino acid to be changed from a nonpolar glycine to a positively charged arginine. This causes receptor activation in the absence of FGF. The result is a condition called achondroplasia, a form of dwarfism. In achondroplasia, chondrocytes differentiate into bone as soon as the mutant FGF receptors are expressed (achondroplasia = no chondrocytes). This usually occurs shortly after birth, and prevents further bone growth. Without chondrocytes, cartilage cannot be produced. This particular form of dwarfism only affects the long bones (e.g., it does not affect the skull). 99% of achondroplasia is caused by dominant alleles for these FGF receptors, and 97% involve the same missense mutation.
FGF signals that the body is finished growing by instructing chondrocytes to differentiate into bone. Usually, the signal is not released until after puberty. However, a single missense mutation on the dominant allele for FGF receptor 3 can cause the 380th amino acid to be changed from a nonpolar glycine to a positively charged arginine. This causes receptor activation in the absence of FGF. The result is a condition called achondroplasia, a form of dwarfism. In achondroplasia, chondrocytes differentiate into bone as soon as the mutant FGF receptors are expressed (achondroplasia = no chondrocytes). This usually occurs shortly after birth, and prevents further bone growth. Without chondrocytes, cartilage cannot be produced. This particular form of dwarfism only affects the long bones (e.g., it does not affect the skull). 99% of achondroplasia is caused by dominant alleles for these FGF receptors, and 97% involve the same missense mutation.
Sex steroids are responsible for maintaining bone density. Rising levels of sex hormones, particularly estrogen, lead osteoblasts to produce bone from epiphyseal cartilage faster than chondrocytes can produce the epiphyseal cartilage, causing cartilage to ultimately disappear. This contributes to the termination of growth during adolescence, and is involved in the process of bones gradually becoming less tolerant to shock. As humans age, they experience a loss of bone mass/density called osteoporosis (porous bones). Osteoporosis occurs more often in females than males.
When high serum calcium is detected, the parafollicular cells (also called C-cells) of the thyroid release calcitonin. Calcitonin binds to inhibitory receptors on osteoclasts. This allows osteoblasts to deposit calcium into bone faster than osteoclasts can remove it, leading to a net increase in bone growth and consequent decrease in blood calcium. Calcitonin also reduces retention of calcium by the kidneys, though this action has a less salient impact on reducing serum calcium.
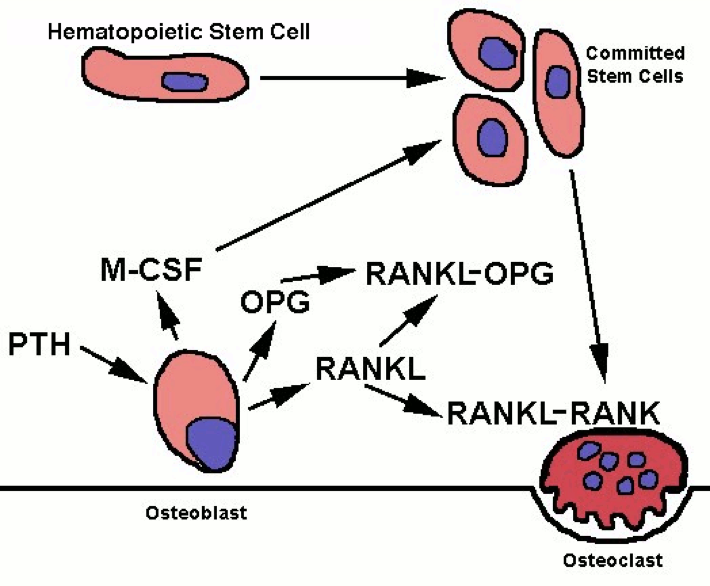 PTH receptor activation has the opposite effect on bone, inducing a shift in activity to favor osteoclasts’ removal of calcium from bone. However, osteoclasts do not express PTH receptors; PTH receptors are found on osteoblasts. Osteoclast precursors and mature osteoclasts express receptor activators of nuclear factor-kappa B (RANK). Activation of RANK promotes osteoclastogenesis, as well as the survival and activity of osteoclasts. The RANK ligand (RANKL) is produced as a paracrine signal by osteoblasts. However, osteoblasts also produce the paracrine signal osteoprotegrin (OPG), which binds to RANKL and prevents its ability to activate RANK. Activation of PTH receptors on osteoblasts enhances the release of RANKL while inhibiting the release of OPG. This enables more RANKL-RANK signaling, increased osteoclast activity, and a consequent increase in osteoclasts’ removal of calcium, which raises blood calcium.
PTH receptor activation has the opposite effect on bone, inducing a shift in activity to favor osteoclasts’ removal of calcium from bone. However, osteoclasts do not express PTH receptors; PTH receptors are found on osteoblasts. Osteoclast precursors and mature osteoclasts express receptor activators of nuclear factor-kappa B (RANK). Activation of RANK promotes osteoclastogenesis, as well as the survival and activity of osteoclasts. The RANK ligand (RANKL) is produced as a paracrine signal by osteoblasts. However, osteoblasts also produce the paracrine signal osteoprotegrin (OPG), which binds to RANKL and prevents its ability to activate RANK. Activation of PTH receptors on osteoblasts enhances the release of RANKL while inhibiting the release of OPG. This enables more RANKL-RANK signaling, increased osteoclast activity, and a consequent increase in osteoclasts’ removal of calcium, which raises blood calcium.
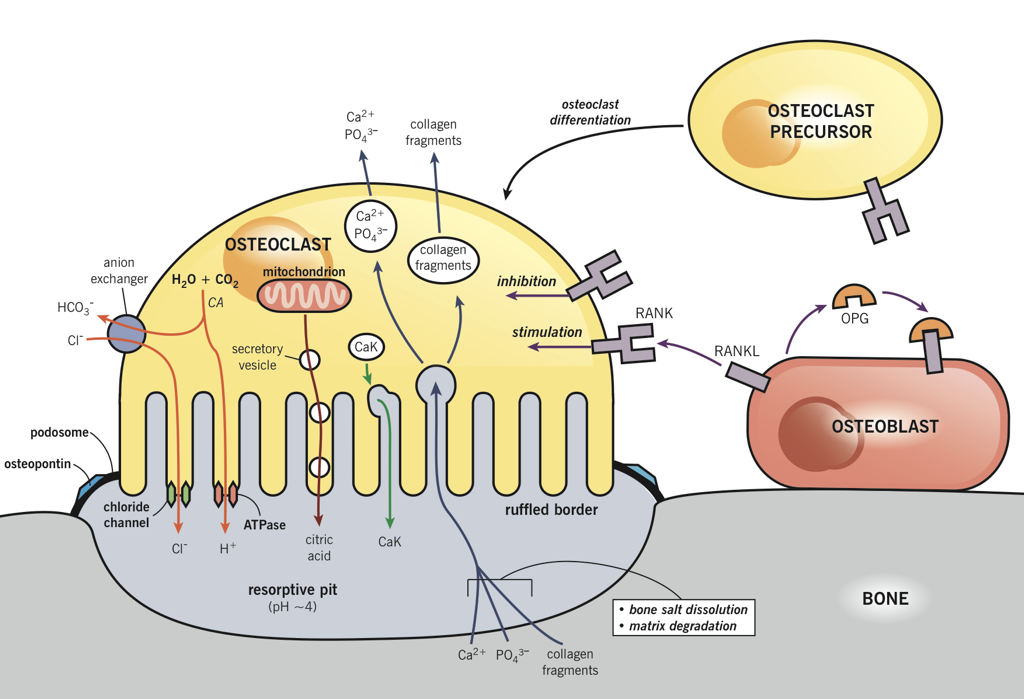
However, the effects of PTH are not limited to bones. PTH also enhances calcium retention by the kidneys (i.e., it decreases the loss of calcium in urine). If there is excessive loss of calcium in the urine (hypercalciuria), high concentrations of calcium can accumulate in the renal pelvis and form kidney stones. In addition to increasing calcium reabsorption by the kidneys, PTH also signals the kidneys to convert 25-hydroxy D3 [25(OH)D] into 25(OH)D, the active form of vitamin D3, which is commonly referred to as calcitriol. Finally, PTH inhibits calcitonin signaling, further potentiating its effects on serum calcium levels.
Calcitriol
Vitamin D3, a lipid, is synthesized in the skin when promoted by UVB light, which promotes 90% of vitamin D synthesis. UVB light catalyzes the conversion(s) of pro-D3 to pre-D3 to vitamin D3, all occurring in the skin. Vitamin D3 can also be consumed in dietary sources, but it must be in the correct form, such as the vitamin D3 in fish oil. The form of vitamin D used to enrich milk is in its primitive form, and still requires conversion by sunlight. UVA light penetrates the skin deeper than UVB rays, and initiates the breakdown of folic acid (vitamin B9) and folate. The lack of folic acid increases the likelihood of birth defects, such as spina bifida, and the lack of folate can cause anemia and slower growth rates.
Vitamin D3 is converted to 25-hydroxy D3 [25(OH)D] by the liver. PTH stimulates the conversion of 25(OH)D by the kidneys to its active form, called calcitriol (1,25-dihydroxycholecalciferol; 1,25-(OH)2D3). Interestingly, calcitriol can also be produced by the liver, and to an even greater extent than by the kidneys. The active calcitriol is able to enhance calcium uptake in the intestines, leading to an increase in serum calcium. However, enzymes rapidly break down calcitriol. This allows for finer control of calcium levels. In a short period of time, calcium concentrations can be dramatically altered. This is particularly important because calcium levels are subject to rapid, dramatic changes, such as loss in urine, and consumption of high-calcium foods.
Vitamin D deficiency can lead to a condition called Rickets, characterized by bowed legs due to weak bones, enlarged joints, muscle cramps, and muscle twitches. However, the addition of vitamin D to milk has largely eradicated Rickets. However, even if vitamin D is present, high quantities of vitamin A can outcompete vitamin D3, leading to calcium deregulation. Vitamin D deficiencies can lead to hypocalcemia. Hypocalcemia is severe when calcium serum concentrations are less than 2.1mMol/L. In addition to vitamin D deficiency, hypocalcemia can result from pancreatitis, hypoparathyroidism, hypercalciuria, and numerous other causes. Hypercalcemia, on the other hand, has two general forms. It can either be due to an increase in calcium, or a moderate decrease in magnesium, which enhances the effects of PTH. Hypercalcemia can lead to overactive skeletal muscles and adipose tissue, and can lead to insulin resistance, type II diabetes, and numerous other complications. The role of the parathyroid in calcium regulation demonstrates the dangers of thyroidectomy, which also removes the parathyroid. Thyroxin deficiency can be managed with synthroid, but calcium regulation is much more challenging to treat.
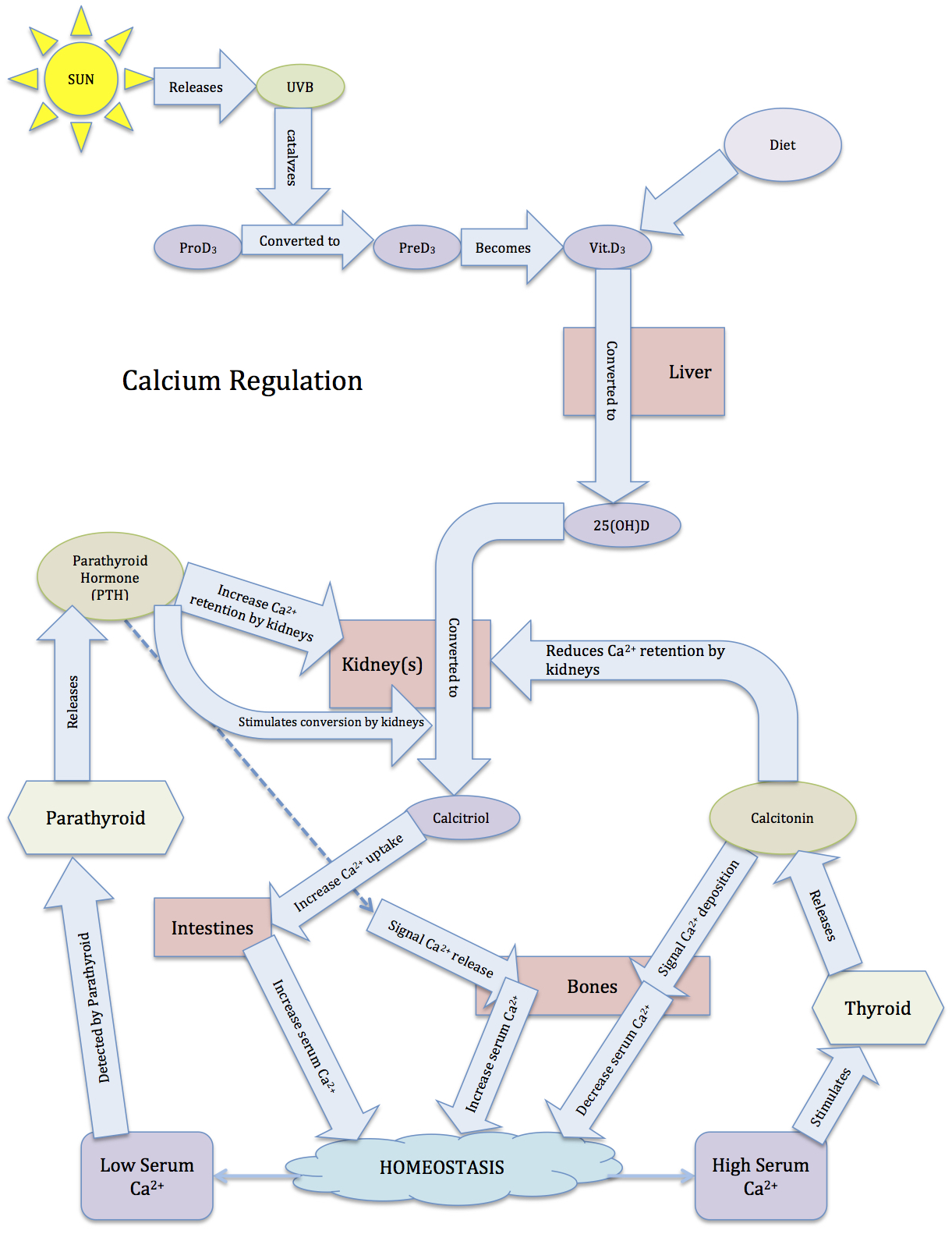
Nevertheless, calcium is only one of many ions whose concentration requires tight regulatory control. The thyroid’s necessity of iodide to produce thyroxin demonstrates the importance of maintaining iodide concentrations, and maintenance of magnesium concentrations is especially important in calcium regulation. Furthermore, other ions, particularly sodium, potassium, hydrogen, and iron, necessitate fine control to maintain homeostasis. Sodium and potassium are involved in almost every aspect of life; when animals evolved out of the salty ocean, they took the ocean with them (including the salt!); humans are largely composed of saltwater. There are beneficial aspects of salt that are necessary for life, but too much, too little, or an imbalance between different ions can have life-threatening consequences. This necessitates tight regulation of serum salt levels.
Feedback/Errata