
1
The Pancreas and Glucose Regulation
The Pancreas
The pancreas has diverse roles in multiple systems: endocrine, paracrine, autocrine, and exocrine. In general, pancreatic functions regulate how food is digested, and how digested food (particularly sugar) is stored and utilized by the body.
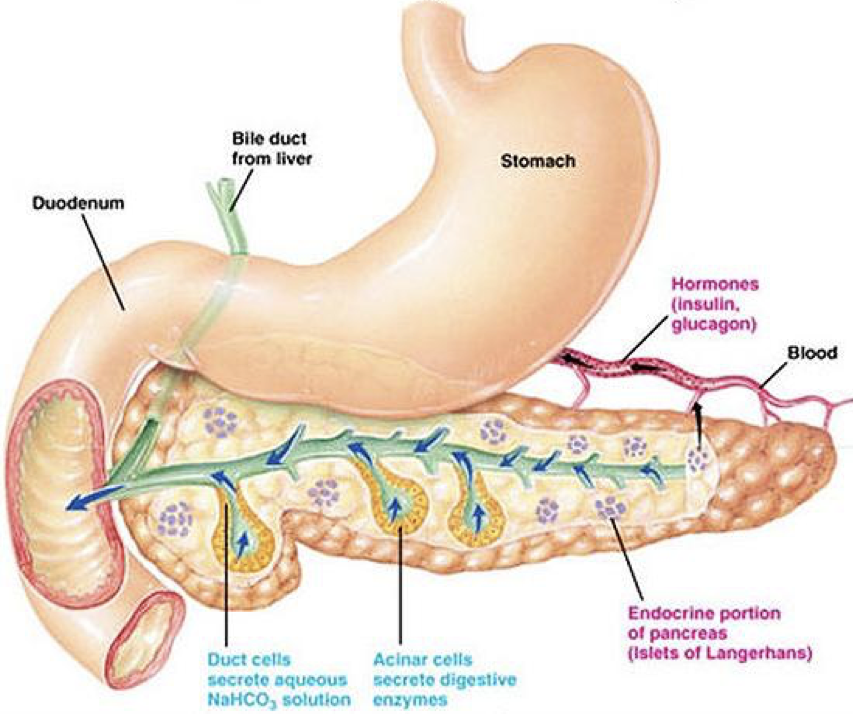
Pancreatic Organization
All mature pancreatic cell types are derived from epithelial cells of the endoderm through a process thought to be mediated by transforming growth factor-β (TGF-β) and fibroblast growth factor (FGF). In humans, the pancreas is actually the combination to two distinct pieces (dorsal and ventral) that converge as they develop. In other animals, such as birds, the dorsal and ventral pancreas remain separate, and have different functions (unless noted otherwise, we’ll be discussing human organs). The singular dorsal pancreas receives directive (i.e., permissive) input from the notochord (of the mesodermal layer) without directly inducing gene expression. The ventral pancreas is divided into two distinct pancreatic buds, and receives developmental input from the lateral plate mesoderm (LPM) but not the notochord. However, this mesodermal input actively induces differentiation of endodermal tissue into pancreatic tissue in the two ventral buds. The two ventral buds become the head of the pancreas, and the dorsal pancreas becomes the body and tail of the pancreas, with the tail reaching towards the spleen and the head wrapped by the duodenum (i.e., the beginning of the small intestine). The entire pancreatic structure is posterior (dorsal) to the stomach. Though the pancreas is largely independent and internally regulated (i.e., without multi-step feedback), its activity is subject to modulation by other systems, such as the autonomic nervous system.
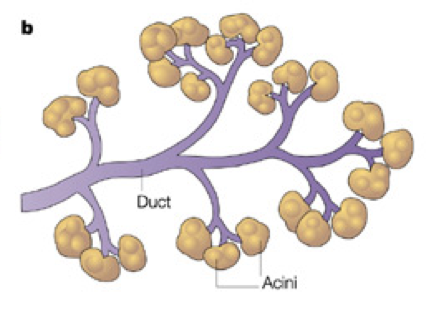
Ductal Cells (duct cells) form a system of excretory ducts throughout the pancreas that allow for excretion into the duodenum of the small intestine via the pancreatic duct. Ductal cells themselves secrete sodium bicarbonate (NaHCO3) into the duodenum to increase the pH. The higher, alkaline pH created by sodium bicarbonate allows lipases to function (remember that different enzymes function optimally at different pH’s; the enzymes in the stomach require a low, acidic pH, whereas the enzymes in the duodenum require a higher, basic pH). Sodium bicarbonate can also be released into the bloodstream to alter pH.
Acinar cells connect to the duct system via centroacinar cells. Acinar cells are organized into glands, sometimes called pancreatic alveoli, which produce digestive enzymes and buffers for excretion into the duodenum. These include amylases for the breakdown of carbohydrates, peptidases (also called proteases or proteolytic enzymes) to break down proteins, nucleases to break down nucleic acids, and lipases to break down lipids. Together, these cells create a matrix that covers most of the pancreas. The system of ducts drains into the pancreatic duct, along with bile from the gallbladder, before draining into the duodenum.
The liver is a giant blood-cleaning organ. Beyond detoxifying the blood, the liver plays many other roles, including numerous endocrine functions. It also picks up excess salts and minerals from the blood stream. When the liver picks up salts and minerals, it selectively stores them in the gallbladder. While the gallbladder is a distinct structure, it is sometimes considered part of the liver. Storing salts in the gallbladder serves two primary purposes: (1) to remove the salts from circulation, and (2) to store them for later use in lipid processing. As such, the gallbladder fills with minerals and salts that can be used at later times within the intestines. However, if there is an over-accumulation of salts and minerals in the gallbladder, there may be a formation of gallstones. Gallstones create a risk of rupturing the gallbladder as they may accumulate into a staggering volume. The treatment of gallstones is often to remove the entire gallbladder. Salts stored in the gallbladder, along with other waste products from the liver, are carried via the bile duct, which converges with the pancreatic duct, before being excreted into the duodenum along with pancreatic excretions.
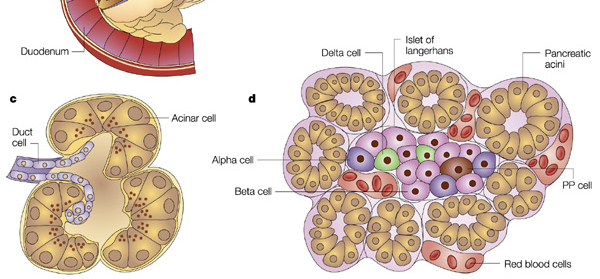
Endocrine secretions of hormones by the pancreas control how digested food is used and stored. Endocrine hormones are synthesized and secreted by islets of Langerhans. Islets of Langerhans (also simply called pancreatic islets) are scattered throughout the exocrine matrix as distinct “islands.” However, they only comprise about 1% of pancreatic tissue, and are only about 2 million in number. Within an islet of Langerhans, there are multiple types of cells that produce hormones.
Cells in the pancreatic islets
Certain cells within pancreatic islets are specialized to measure serum glucose levels. If the serum glucose levels are high, insulin is secreted by pancreatic β cells (whichThe suffix –emia means “in-blood” or refers to the presence of a substance in blood.
compose 60% of islet cells). Insulin signals adipocytes (fat cells), myocytes (muscle cells), and hepatocytes (liver cells) to uptake glucose from the blood stream. If β cells fail to respond to increases in serum glucose levels, hyperglycemia (high blood sugar) will result.
Aside from cells in the brain, all cells in the body are controlled by insulin to a certain degree. The brain runs on glucose, and is the primary user of sugar within the body. However, brain cells do not require insulin to uptake glucose. This lack of insulin regulation of the brain’s uptake of glucose is vital. The brain always demands a large amount of glucose, so if brain cells relied on insulin to govern their glucose uptake, brain functioning would undergo dramatic fluctuations. In other words, glucose uptake by brain cells is insulin-independent.
If the blood glucose levels are low, glucagon is secreted by pancreatic α-cells (composing 25% of islet cells), which causes the liver to release glucose into the blood to raise blood sugar. Failure of this α-cell response results in hypoglycemia, low blood sugar.
Pancreatic polypeptide cells (PP cells) release pancreatic polypeptide (PP). PP is released in two waves to shut down digestive processes: first when food is in the stomach, and second when food enters the duodenum. PP is produced when high concentrations of protein are present, which often occurs when the body is low on sugar. This is a common environment after a protein-rich meal, during fasting, while exercising, and in the event of acute hypoglycemia. PP’s primary role is paracrine; it regulates the release of other pancreatic hormones. However, the effects of PP on α and β cell functioning can vary. PP release is decreased by somatostatin and intravenous glucose. Elevated PP levels exert an appetite suppressing effect through its interactions with the hypothalamus, and PP may play a causal role in the development of anorexia nervosa (PP is often elevated in anorexic patients). Its close link to leptin (a long-term signal of satiety) provides a further hint at its possible role in anorexia.
δ cells (comprising 10% of islet cells) produce somatostatin (SST; also called growth hormone inhibitory hormone, GHIH). SST released from pancreatic δ cells acts in a paracrine manner to suppress the release of other pancreatic hormones (e.g., SST inhibits 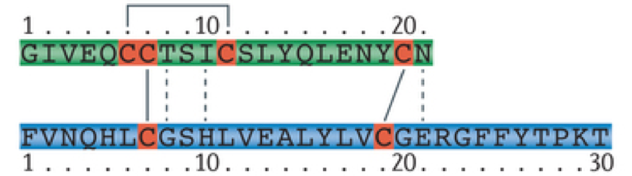
Prandial is derived from the Latin word for “meal.”
release of PP). However, SST is also released from other regions of the body (e.g., the stomach, duodenum, and hypothalamus). SST released from the hypothalamus mediates negative feedback by growth factors such that growth factors activate the release of SST, which inhibits the release of more growth factors by the anterior pituitary.
ε cells produce ghrelin, which, in contrast to PP, signals appetite and regulates gastrointestinal function, nutrient storage, and nutrient utilization. Islets are organized such that somatostatin- and glucagon-releasing cells are in the periphery, encasing insulin-releasing cells that are concentrated in the centers of islets. Importantly, there are not very many cells within each pancreatic islet, and they do not reproduce.
Glucose Regulation
Most organisms use glucose as a source of energy. Glucose is the only source of fuel used in the brain (with a few exceptions resorted to only under dire circumstances, such as ketone bodies). The normal protocol is for the brain to receive sugar first, and then the brain will grant permission to other regions of the body to receive sugar. Blood glucose generally fluctuates between about 70 and 99 mg/dl prior to eating, and rises to about 140 mg/dl after eating (post-prandial). There is about 450-500 grams of glucose in the body. The brain is always in high demand of glucose, and uses about 125 grams per 24-hour period. However, under heightened levels of activity (e.g., during a final exam), the brain can use as much as a pound (~450g) in just a few hours! In the two hours before and the two hours after eating, insulin and glucagon have a tug-of-war to manage high or low blood glucose levels, respectively. Both hormones operate constantly without true negative feedback.
Because of the regular fluctuations in blood sugar, instantaneous blood sugar is not a reliable measure. Rather, glycated hemoglobin (HbA1c, or just A1c) can be measured to determine blood sugar averages over long periods of time (up to about three months). The higher the concentration of glucose in the bloodstream, the more glucose molecules bind to hemoglobin; these glucose molecules remain permanently bound for the duration of the red blood cell’s lifespan. As a result, the amount of glycated hemoglobin in a red blood cell reflects that cell’s average degree of exposure to glucose over the course of its approximately four-month life cycle. Normal HbA1c levels should remain lower than about 5.7%.
Due to the importance of blood sugar homeostasis, the two pancreatic hormones, insulin and glucagon, are particularly important.
Insulin
Insulin is an anabolic peptide hormone. It is hydrophilic and cannot diffuse through membranes. Insulin is composed of a 21 amino-acid alpha chain and a 30 amino-acid beta strand. In its natural form, insulin can only exist in the body for a brief period, having a half-life of only about 4-6 minutes. This is because there are numerous enzymes in the body that break down endogenous insulin. This poses a significant challenge when administering insulin as a treatment for hyperglycemia. In the past, insulin has been harvested from pigs for use in humans with insulin deficiencies (unfortunately this causes an allergic reaction for some people). However, after having sequenced the human genome, it has been possible to repurpose beer breweries to mass-produce insulin using bacteria, and to modify the molecular structure of insulin to prevent rapid degradation, extending the duration of action. The rapid degradation of insulin also poses challenges for measuring levels of insulin in the bloodstream. However, a biproduct of insulin production called the free C peptide remains in the bloodstream for a longer period of time and provides a reliable indicator of levels of insulin production.
Insulin exerts its effects throughout the body. It increases glucose uptake in muscle cells and fat cells, and increases glycogenesis (creation of glycogen) in the liver and muscle cells while simultaneously decreasing glycogenolysis (breakdown of glycogen). It also increases amino acid uptake and protein synthesis, increases fatty acid synthesis and storage (e.g., in adipose tissue), and strongly promotes satiety through interactions in the hypothalamus. Furthermore, insulin increases the utilization of glucose by promoting glycolysis to generate ATP. Finally, insulin also has an important effect on β cells themselves. β cells release insulin as an autocrine signal. β cells express pro-apoptotic factors. However, these pro-apoptotic factors are rendered inert through phosphorylation. The binding of insulin to autoreceptors on β cells activates protein kinases that phosphorylate the pro-apoptotic factors, which prevents their degradation of β cells and enables healthy β cell functioning. If insulin auto-stimulation is hampered, the consequent reduction in phosphorylation of pro-apoptotic factors can damage β cells.
Glucose Transporters
Importantly, different cells have different types of glucose transporters, which allow glucose to enter cells under different conditions. GLUT1 is the primary glucose transporter, and resides on most cells in the body. However, GLUT1 is not very prevalent on cells, and is relatively inefficient because its transport of glucose depends on the concentration of glucose. GLUT2 operates independent of insulin, and is largely expressed in pancreatic β cells, though it is also expressed in the hypothalamus, liver, and even the kidneys.
GLUT3 is found in the brain and gonads, and like GLUT2, is independent of the actions of insulin. Not only is its operation independent of insulin, but GLUT3 also transports glucose in a glucose-concentration-ind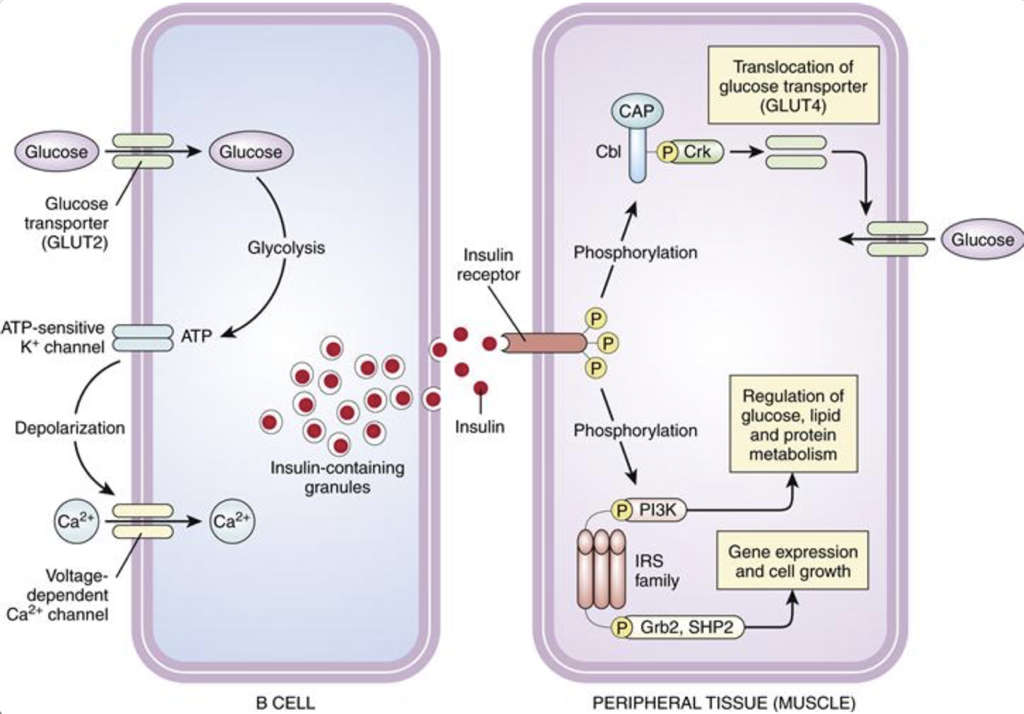 ependent fashion. This is beneficial because the cerebrospinal fluid has a lower sugar concentration than blood, so it is vital that the GLUT3 transporter be efficient in order to adequately fuel the brain. GLUT4, in contrast, is dependent on the actions of insulin. It is found in muscles, adipose tissue, and to a minor degree in the hypothalamus. When translocated to the membrane in response to insulin signaling, GLUT4 dramatically enhances the influx of glucose.
ependent fashion. This is beneficial because the cerebrospinal fluid has a lower sugar concentration than blood, so it is vital that the GLUT3 transporter be efficient in order to adequately fuel the brain. GLUT4, in contrast, is dependent on the actions of insulin. It is found in muscles, adipose tissue, and to a minor degree in the hypothalamus. When translocated to the membrane in response to insulin signaling, GLUT4 dramatically enhances the influx of glucose.
Insulin Release
Pancreatic β cells determine that insulin secretion is necessary when there is a rise in the presence of glucose. Glucose is transported into β cells by GLUT2 along its concentration gradient. As a result, the amount of glucose entering β cells indicates the concentration of glucose in the bloodstream. Once glucose enters β cells, it is converted into ATP. Accumulating ATP closes ATP-sensitive potassium channels. Normally, these ATP-sensitive potassium channels are open. Since β cells have a high internal concentration of potassium relative to the external concentration, potassium will flow out of β cells while these channels are open, carrying a positive charge out of the cell, and increasing the membrane polarity (i.e., membrane polarity hyperpolarizes towards the equilibrium potential of potassium). When ATP closes these potassium channels, the outward flow of potassium is blocked. Without the flow of potassium out of the β cell, the membrane will become less polar (i.e., the membrane will depolarize). The depolarization resulting from the closure of ATP-sensitive potassium channels alters the conformation of voltage-gated calcium channels, opening their lumen and permitting an influx of calcium into the cell (calcium is much more concentrated outside of β cells than inside β cells). The influx of calcium initiates the exocytotic release of insulin stored in vesicles. Since only the influx of calcium is necessary to initiate insulin release, any event that depolarizes β cells towards calcium channel thresholds can also drive the release of insulin. For example, when there is an increase in serum potassium (hyperkalemia), there is a decreased efflux of potassium from β cells, which causes depolarization. This leads to insulin release, and may ultimately lead to hypoglycemia.
Interestingly, Endocrine regulation by β cells is not limited to the vital roles of insulin. Amylin is co-secreted with insulin. Amylin inhibits glucagon secretion, delays gastric emptying, and acts as a satiety signal to the brain.
 there are certain molecules that are similar enough to glucose that they will enter through GLUT2 (but not GLUT4) and drive the production of ATP. For example, glucosamine can be transported through GLUT2 on β cells and drive the production of ATP and subsequent release of insulin.
there are certain molecules that are similar enough to glucose that they will enter through GLUT2 (but not GLUT4) and drive the production of ATP. For example, glucosamine can be transported through GLUT2 on β cells and drive the production of ATP and subsequent release of insulin.
Insulin Effects
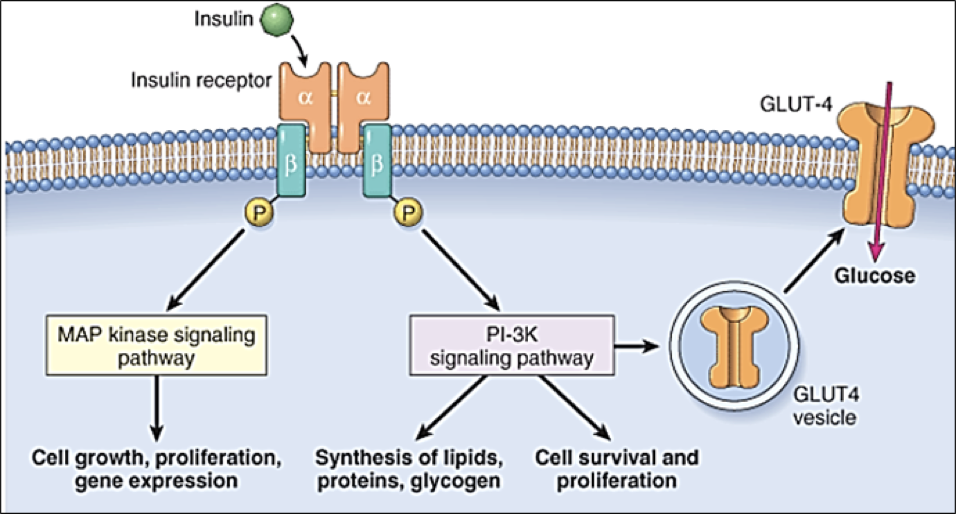
Insulin receptors are found on almost every cell outside of the nervous system. They are homomeric dimers, composed of two identical quaternary proteins, each of which has an alpha subunit and a beta subunit (for a total of two alpha and two beta subunits). Each insulin receptor alpha subunit, exposed on the exterior of the cell, must be bound by insulin; two insulin molecules must bind to the insulin receptor. Upon stimulation by two insulin molecules, insulin receptor complexes initiate multiple signaling pathways.
Firstly, insulin receptor activation promotes the translocation of glucose transporter 4 (GLUT4) into the membrane, permitting glucose transport into the cell. This same pathway also promotes lipid, protein, and glycogen synthesis. However, insulin receptor activation also promotes cell growth, proliferation, and gene expression through an MAP kinase signaling pathway. Importantly, some molecules other than insulin may bind to insulin receptors.
Glucagon
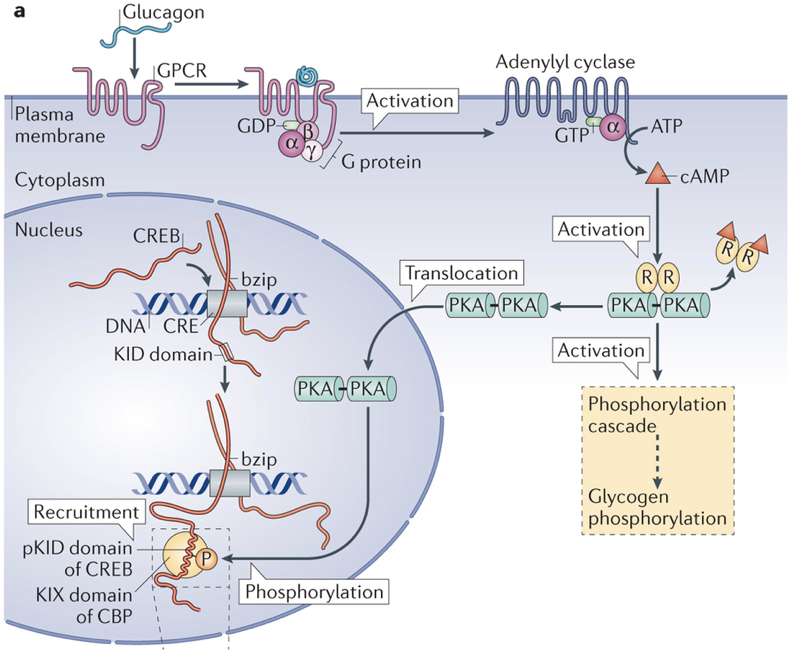
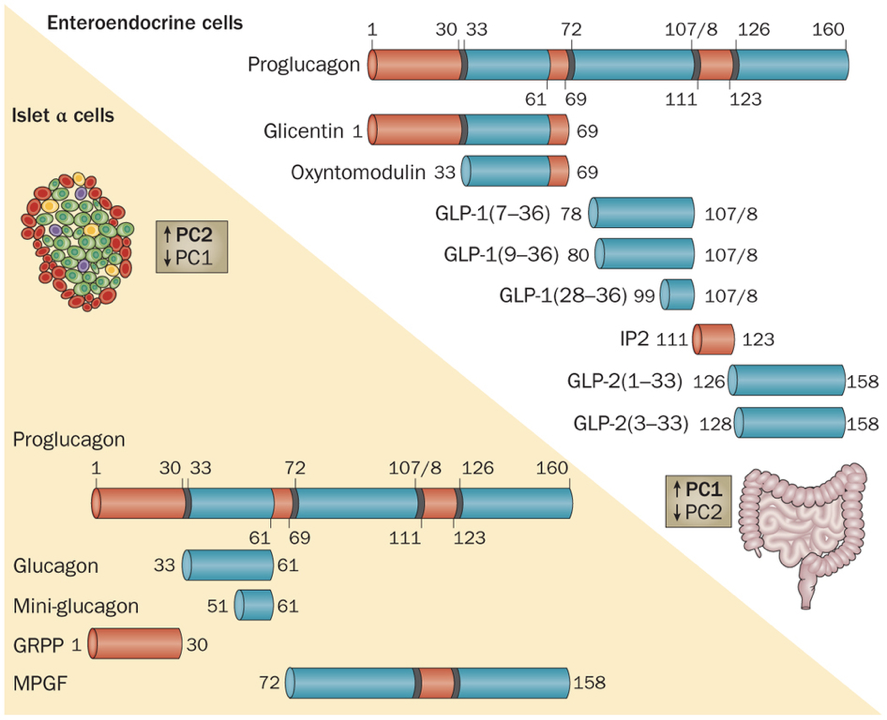 Like insulin, glucagon is also a peptide hormone. It is only composed of 29 amino acids, and is produced by the cleavage of a middle section of proglucagon, a large 160 amino acid protein. Glucagon exerts its effects through a G-protein-coupled receptor, activation of which enables adenylyl cyclase to convert ATP to cyclic AMP (cAMP). The increased production of cAMP leads to the phosphorylation of glycogen, and subsequent release of glucose. Additionally, glucagon also initiates alterations in gene transcription.
Like insulin, glucagon is also a peptide hormone. It is only composed of 29 amino acids, and is produced by the cleavage of a middle section of proglucagon, a large 160 amino acid protein. Glucagon exerts its effects through a G-protein-coupled receptor, activation of which enables adenylyl cyclase to convert ATP to cyclic AMP (cAMP). The increased production of cAMP leads to the phosphorylation of glycogen, and subsequent release of glucose. Additionally, glucagon also initiates alterations in gene transcription.
Also like insulin, glucagon has a variety of effects. Some of its primary effects are on the liver, where it enhances glucose output (release), lipid oxidation, and cell survival, while also decreasing the rate of lipid synthesis. This is of particular importance because the liver is one of the primary energy storage sites, where glucose is stored in the form of glycogen. Adipose tissue (specifically white adipose tissue) is another energy storage site in the body, and glucagon enhances lipolysis and thermogenesis in such cells. In the heart, glucagon increases heart rate and lipid oxidation, albeit at the expense of cardiac myocyte survival. In the gastrointestinal tract, glucagon decreases motility, permitting greater absorption of nutrients, and less expenditure of energy. In the kidneys, glucagon increases the glomerular filtration rate and water reabsorption. Part of the sympathetic response is actually to stimulate α cell release of glucagon to provide a rise in blood glucose necessary to fuel fight-or-flight decision-making and behavior. This sympathetic nervous system-induced release of glucagon is independent of PP. Like insulin, glucagon affects appetite at the level of the brain. However, these effects of glucagon are extremely context-dependent.
One of the primary roles of the pancreas is to maintain blood glucose within a discrete range (i.e., to maintain homeostasis). However, despite being largely autonomous, the pancreas can be modulated such that the discrete range, within which the pancreas strives to preserve blood sugar levels, can be up- or down-regulated by the autonomic nervous system.
As it turns out, the pancreas receives innervation by midbrain structure projections (e.g., the vagus nerve), as well as the splanchnic nerve, and by the intermediolateral nucleus via the coeliac ganglion. Overall, it may not be quite as independent as has previously been thought. The brain can actually override the pancreas so that it can meet its high demand for glucose.
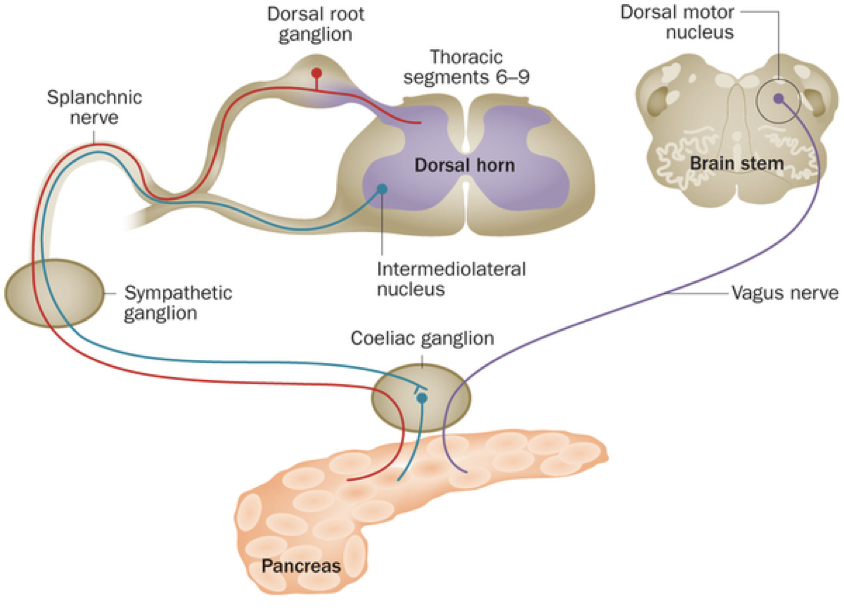
Furthermore, evidence indicates that paracrine signaling within pancreatic islets serves an important role within blood glucose regulation. In fact, 40-50% of β cells are in direct contact with α cells, enabling efficient paracrine signaling. Insulin has been shown to directly inhibit the secretion of glucagon. However, β cells are also responsive to glucagon, which binds to receptors coupled to adenylyl cyclase. α cells are the first islet cells to develop, and their glucagon signaling pathways are vital for the early differentiation of β cells. The beneficial effects of glucagon on β cells continue throughout the lifespan. These beneficial effects generally relate to β cell glucose competence: their ability to respond efficiently to various levels of glucose. β cells adjacent to α cells tend to receive more paracrine glucagon signals, and exhibit better glucose competence, compared to β cells more distant from α cells. Furthermore, glucose competence can be restored in poorly responding β cells with the administration of glucagon to those cells. These beneficial effects of glucagon on β cell functioning are due to glucagon signaling pathways that lead to the phosphorylation of CREB in β cells, which controls the expression of proinsulin and anti-apoptotic factors. This may seem contradictory because glucagon has hyperglycemic effects, while promoting the hypoglycemic effects of insulin. However, this apparent contradiction is resolved by the cosecretion of miniglucagon with glucagon. About 5% of glucagon in α cells is cleaved into miniglucagon prior to release. Miniglucagon signaling in β cells prevents glucagon from stimulating insulin secretion without preventing glucagon’s beneficial effects on β cell glucose competence. In addition to its paracrine role within pancreatic islets, miniglucagon also has an interesting endocrine role. Like insulin, miniglucagon promotes translocation of GLUT4, enhancing glucose uptake. While the two hormones initiate this process through different receptor interactions, the two pathways converge from then on, such that miniglucagon and insulin share the final steps of GLUT4 translocation. This is particularly interesting because it enables miniglucagon to exert the same effect as insulin on GLUT4 translocation while bypassing insulin receptors.
Diabetes means “to go through,” or “siphon,” and mellitus means “sweet,” or “honey,” so diabetes really just means “sweet blood.”
Diabetes Mellitus
Deficiencies in the regulatory mechanisms for blood sugar can result in diabetes mellitus (DM), which is characterized by high blood sugar.
Diabetes is the number one endocrine disorder, though metabolic syndrome is also prevalent and often comorbid with diabetes. When blood sugar is high, hyponatremia (low blood sodium) can result from excessive urination. As sugar leaves the kidneys, water follows it, and sodium follows the water. The result is lower-than-normal blood sodium. Diabetes can also lead to polyphagia, which is excessive hunger or appetite, resulting largely from the absence of insulin’s appetite suppressing effects on the hypothalamus. Ketonuria is the excessive excretion of ketone bodies by the kidneys, and is caused by excessive protein breakdown (e.g., from the muscles), which are a backup energy source when glucose is depleted. Due to the stress placed on the kidneys, diabetic nephropathy can result from damage to renal blood vessels, in particular, the glomeruli. Diabetic retinopathy, a progression of vision problems that can ultimately lead to blindness, is caused by damage to retinal blood vessels. Lastly, diabetic neuropathy can result from damage to nerves (most often in the legs and feet).
Type I Diabetes Mellitus
Type I diabetes mellitus (T1DM) is the result of a failure to release insulin. It is theoretically presumed to result from autoimmune targeting of beta cells, though this remains controversial. Modern theories suggest that the cause of T1DM may be viral.
During development, pancreatic progenitor cells differentiate into either endocrine or exocrine cells. Diabetes type I results if there are no insulin-producing β cells. Most commonly, diabetes 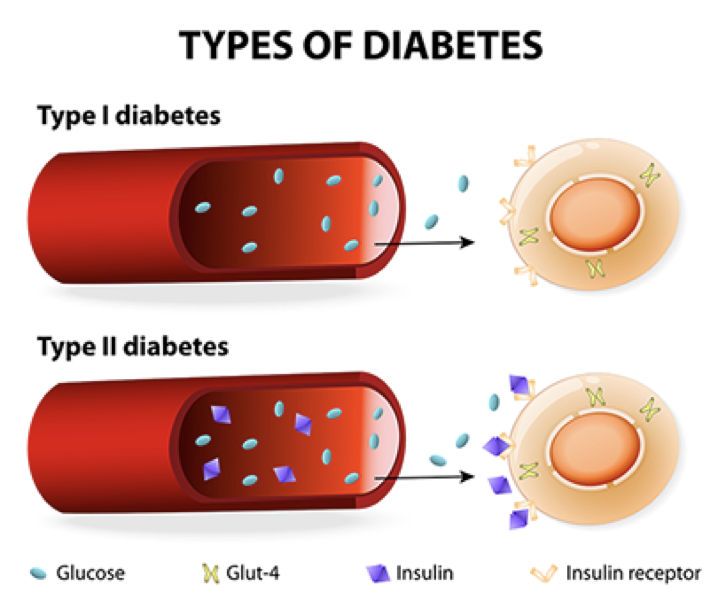 type I is due to a lack of pancreatic β cells that produce and secrete insulin. However, lack of sugar receptors on β cells, lack of a release mechanism in functional β cells, or a failure to produce insulin in β cells can also result in a failure to release insulin. The consequence of type I diabetes is an inability to efficiently respond to high blood sugar. Diabetes type I can occur before birth, during childhood, or in puberty. However, when there is a loss of insulin-producing b cells in adults, the resulting condition is denoted as diabetes type 1.5, or late autoimmune diabetes of adults
type I is due to a lack of pancreatic β cells that produce and secrete insulin. However, lack of sugar receptors on β cells, lack of a release mechanism in functional β cells, or a failure to produce insulin in β cells can also result in a failure to release insulin. The consequence of type I diabetes is an inability to efficiently respond to high blood sugar. Diabetes type I can occur before birth, during childhood, or in puberty. However, when there is a loss of insulin-producing b cells in adults, the resulting condition is denoted as diabetes type 1.5, or late autoimmune diabetes of adults 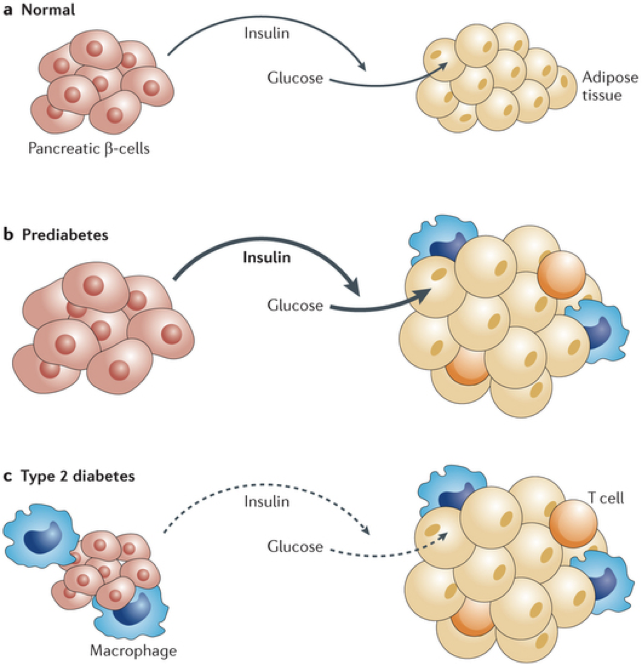 (LADA). Due to the co-secretion of amylin with insulin, the loss of β cells in diabetes type I also entails a loss of amylin. Among other things, this causes food to move through the intestines more rapidly, and removes the satiety-promoting effects of amylin. The lack of amylin has a significant impact on type I diabetics. This also explains why treatment with just insulin fails to resolve all complications, even when the provision of supplementary insulin is tightly regulated.
(LADA). Due to the co-secretion of amylin with insulin, the loss of β cells in diabetes type I also entails a loss of amylin. Among other things, this causes food to move through the intestines more rapidly, and removes the satiety-promoting effects of amylin. The lack of amylin has a significant impact on type I diabetics. This also explains why treatment with just insulin fails to resolve all complications, even when the provision of supplementary insulin is tightly regulated.
Type II Diabetes Mellitus
Type II diabetes mellitus (T2DM) is characterized by a lack of insulin receptor sensitivity. Though there is functional release of insulin (albeit less than normal), insulin receptors are insensitive and fail to detect the presence of insulin, resulting in a failure to respond to high blood sugar. Before developing type II diabetes, patients progress through a pre-diabetic state in which there is an initial rise in insulin secretion, which is most commonly precipitated by a high-calorie diet. This increased demand for and release of insulin causes both β cells and their targets to become hypertrophic. The target cells stop producing functional insulin receptors, and become resistant to insulin’s effects, a state called insulin resistance, or insulin insensitivity. Additionally, the target cells become more susceptible to phagocytes and other immune cells. Without functional insulin receptors, even more insulin is required to elicit the same effect, placing an even higher demand on β cells. As β cells are overexerted, they atrophy. Thus, in diabetes type II, there is a stage of pre-diabetes in which there is an initial rise in insulin production and glucose uptake, followed by the development of insulin resistance and β cell atrophy, and culminating in both a diminished production of insulin, and a diminished response to what little insulin remains.
Because high blood sugar can precipitate T2DM, deregulation of internal signaling pathways driving hyperglycemia can also precipitate T2DM. For example, overproduction of glucagon can raise blood sugar, and chronic stress that increases circulating glucocorticoids also raises blood sugar (while simultaneously inhibiting the hypoglycemic actions of insulin); both can lead to the development of T2DM.
Obesity and Diabetes
Obesity is a primary risk factor for the development of 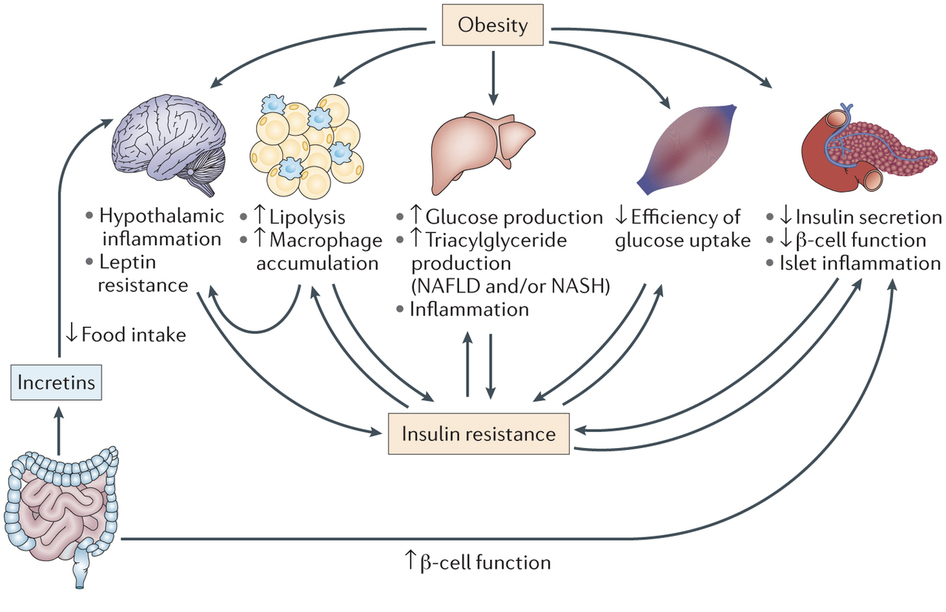 diabetes type II. Obesity has a variety of effects that promote insulin resistance, including hypothalamic inflammation, leptin resistance, increased lipolysis and macrophage accumulation in adipose tissue, increased glucose and triglyceride production in the liver, and decreased β cell function. Importantly, the mesentery, which is largely composed of adipose tissue, is enlarged as a consequence of obesity. The accumulation of adipose tissue drives internal cytokine and chemokine signaling that causes insulin-signaling defects and produces low-grade systemic inflammation. Overall, there are a lot of factors driving insulin resistance in obesity. However, one important and relatively common cause of diabetes type II is a gluconoma, a small tumor in the islets of Langerhans that specifically affects α cells. This can lead to excessive glucagon production, which raises blood sugar. The increased blood sugar promotes increased insulin secretion, which leads to the development of insulin resistance.
diabetes type II. Obesity has a variety of effects that promote insulin resistance, including hypothalamic inflammation, leptin resistance, increased lipolysis and macrophage accumulation in adipose tissue, increased glucose and triglyceride production in the liver, and decreased β cell function. Importantly, the mesentery, which is largely composed of adipose tissue, is enlarged as a consequence of obesity. The accumulation of adipose tissue drives internal cytokine and chemokine signaling that causes insulin-signaling defects and produces low-grade systemic inflammation. Overall, there are a lot of factors driving insulin resistance in obesity. However, one important and relatively common cause of diabetes type II is a gluconoma, a small tumor in the islets of Langerhans that specifically affects α cells. This can lead to excessive glucagon production, which raises blood sugar. The increased blood sugar promotes increased insulin secretion, which leads to the development of insulin resistance.
Treatment of Diabetes
Insulin analogs
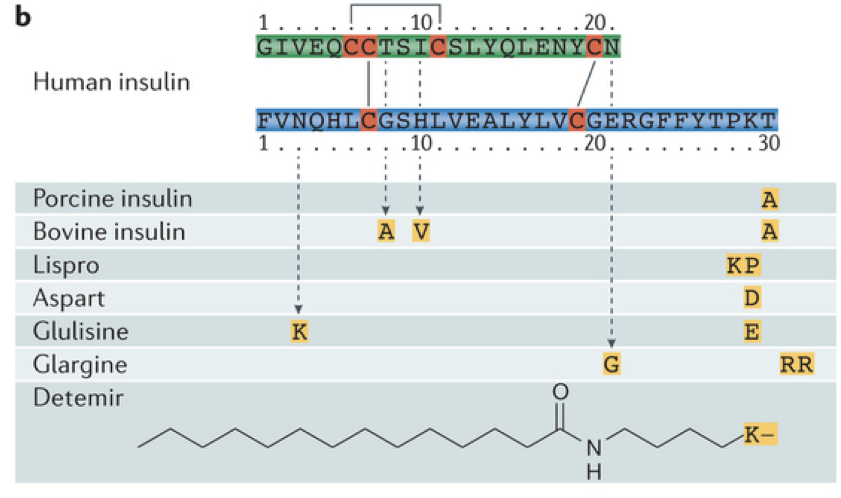
Because insulin itself is subject to rapid degradation, insulin analogs have emerged as a more promising treatment for diabetes because they are not broken down as rapidly. Furthermore, insulin analogs can be tailored for specific purposes based on their duration of action. Two examples are Glulisine and Glargine. Glulisine is fast acting, whereas glargine is long acting.
Incretins
Another promising therapeutic agent is the incretin glucagon-like peptide 1 (GLP-1), which is synthesized by cleaving a portion of proglucagon that neighbors the glucagon sequence. GLP-1 is produced by enteroendocrine cells in response to the presence of food in the intestinal tract. GLP-1 inhibits the release of glucagon by α cells (likely mediated through effects on SST secretion) while enhancing the glucose-dependent release of insulin by β cells and increasing insulin receptor sensitivity. GLP-1 binding to receptors on β cells increases the activity of adenylyl cyclase, yielding an increase in cAMP production. Downstream effects include alterations in gene transcription that increase the production of insulin as well as alterations in ion channels that promote calcium influx. Together, these effects yield a heightened insulin response to a given concentration of glucose.
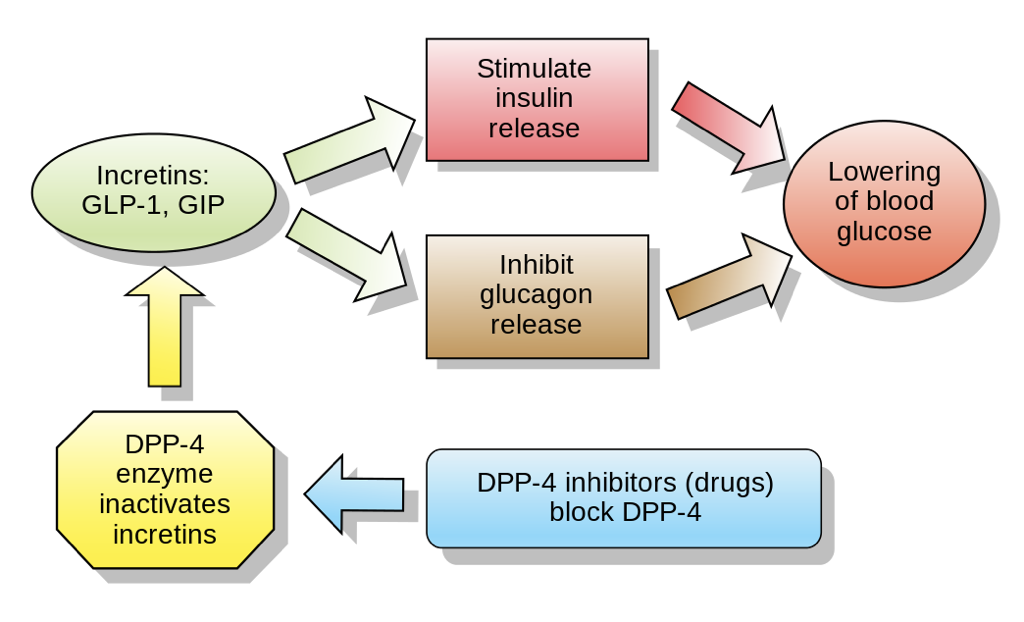 GLP-1 also modulates the brain’s control of the pancreas, and promotes satiety through interactions with the hypothalamus. Nevertheless, GLP-1 has a half-life of only two minutes due to its rapid degradation by enzymes (primarily dipeptidyl peptidase 4, DPP-4) and due to its clearance by the kidneys. For this reason, DPP-4 inhibitors also show promise as therapeutic agents for the treatment of hyperglycemic disorders such as type II diabetes. GLP-1 also acts as a neurotransmitter, and is produced by neurons in the nucleus of the solitary tract and reticular nucleus of the medulla oblongata. DPP-4 is a more promising treatment for T2DM than GLP-1 itself because of its ability to enhance the functioning of an endogenous system, as opposed to trying to replicate the functioning of an endogenous system. Furthermore, by enhancing the endogenous insulin response, the risk of hypoglycemia following excessive insulin administration is significantly mitigated. GLP-1 analogs are also emerging as a viable and effective treatment option.
GLP-1 also modulates the brain’s control of the pancreas, and promotes satiety through interactions with the hypothalamus. Nevertheless, GLP-1 has a half-life of only two minutes due to its rapid degradation by enzymes (primarily dipeptidyl peptidase 4, DPP-4) and due to its clearance by the kidneys. For this reason, DPP-4 inhibitors also show promise as therapeutic agents for the treatment of hyperglycemic disorders such as type II diabetes. GLP-1 also acts as a neurotransmitter, and is produced by neurons in the nucleus of the solitary tract and reticular nucleus of the medulla oblongata. DPP-4 is a more promising treatment for T2DM than GLP-1 itself because of its ability to enhance the functioning of an endogenous system, as opposed to trying to replicate the functioning of an endogenous system. Furthermore, by enhancing the endogenous insulin response, the risk of hypoglycemia following excessive insulin administration is significantly mitigated. GLP-1 analogs are also emerging as a viable and effective treatment option.
Another incretin, Gastric Inhibitory Peptide (GIP), has effects similar to GLP-1. We will return to GIP when discussing gastric hormones.
Miniglucagon is emerging as another promising treatment for T2DM. The rationale emerges from miniglucagon’s effects on GLUT4 translocation, which bypass insulin receptors that may be compromised due to insulin resistance in T2DM.
Feedback/Errata