
3 Endocrine Regulation of Blood Pressure & Volume
Endocrine Regulation of Blood Pressure and Volume
One vital role of the endocrine system is to respond to conditions that alter the body’s ability to efficiently deliver oxygen and nutrients to cells throughout the body while removing carbon dioxide and other waste products. To accomplish these tasks, the cardiovascular system must efficiently circulate blood: through the pulmonary system, where oxygen and carbon dioxide are exchanged through pulmonary respiration; through the hepatic and renal systems, where blood is processed and filtered, respectively; and to all other cells throughout the body, which require the delivery of nutrients and oxygen, and the removal of carbon dioxide and other waste products.
The efficient circulation of blood throughout the body by the cardiovascular system requires adequate blood pressure. Blood pressure can be regulated by cardiovascular changes, such as vasoconstriction and vasodilation, and by changes in cardiac output. However, establishing and maintaining appropriate blood pressures also depends on the volume of blood in circulation. Blood volume can be modulated by changes in plasma volume and by changes in hematocrit volume (e.g., by changing the number of red blood cells). Changes in plasma volume are largely accomplished through changesEndWG08
in the volume of water and the concentrations of salts. Furthermore, there must be an adequate number of red blood cells to carry oxygen and carbon dioxide (i.e., changing the number of red blood cells is necessary to regulate oxygen carrying capacity). All of these factors governing the efficiency of blood circulation are subject to regulation by the endocrine system.
To establish and maintain appropriate blood pressures, the endocrine system must be able to respond to both decreases and increases in blood pressure. Short-term changes in blood pressure are accomplished through changes in vasodilation and vasoconstriction, but longer-term changes in blood pressure require changes in blood volume. When cardiovascular changes fail to accommodate longer-term changes in blood pressure, there is a mid-term endocrine response that changes the plasma volume. Finally, long-term changes in blood volume and oxygen carrying capacity may be necessary to accommodate changing environmental demands (e.g., changes in altitude that entail changes in atmospheric oxygen concentrations). The endocrineEndWG09
system also has a long-term response to changes in blood pressure that involve changing the number of red blood cells (hematocrit volume), which consequently changes the oxygen carrying capacity of blood.
To begin, we will cover the basics of the short-term, mid-term, and long-term endocrine responses to decreasing blood pressure/volume. Then, we will cover the mid-term and long-term responses to decreased blood pressure in depth.
Overview of Responses to Decreased Blood Pressure/Volume
Short-Term Responses to Decreased Blood Pressure/Volume
The short-term response to a decrease in blood pressure/volume (or a need for higher blood pressure and higher oxygen-carrying capacity) is to increase cardiac output and peripheral vasoconstriction. This is accomplished by the release of norepinephrine and epinephrine under control by the sympathetic nervous system. In a normal person, cardiac output can be quadrupled. However, some athletes can raise their cardiac output by a factor of six to ten. Unfortunately, this is only a short-term response thatEndWG10
increases blood pressure without changing blood volume and is not sustainable over an extended period of time. Longer-term responses to decreased blood pressure entail changes in blood volume.
Mid-Term Responses to Decreased Blood Pressure/Volume
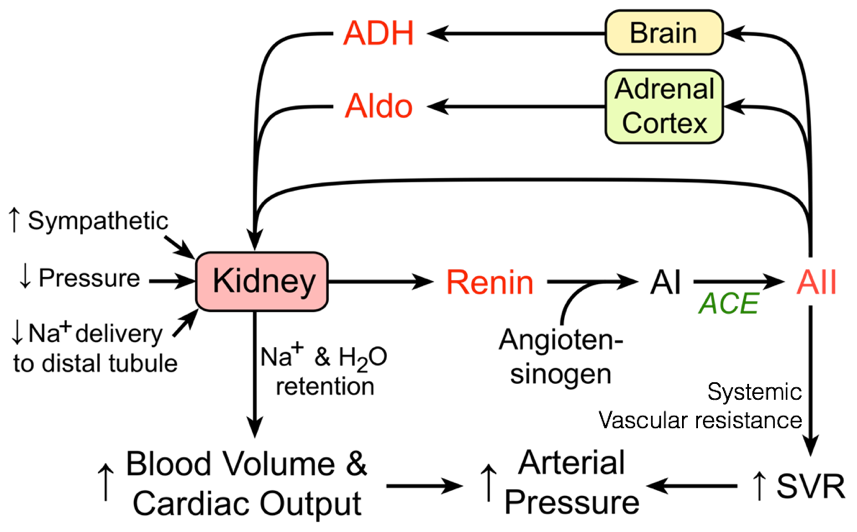 Mid-term responses to decreased blood pressure/volume are accomplished through changes in the volume of blood plasma (i.e., by changing the volume of water and concentrations of salts). These changes in the volume of blood plasma are accomplished primarily through the renin-angiotensin-aldosterone system (RAAS), which also incorporates antidiuretic hormone (also called vasopressin).
Mid-term responses to decreased blood pressure/volume are accomplished through changes in the volume of blood plasma (i.e., by changing the volume of water and concentrations of salts). These changes in the volume of blood plasma are accomplished primarily through the renin-angiotensin-aldosterone system (RAAS), which also incorporates antidiuretic hormone (also called vasopressin).
In response to decreased blood pressure, the kidneys produce and release renin. Once released, renin converts circulating angiotensinogen into angiotensin I. Angiotensin converting enzyme, found in the lungs, converts angiotensin I into angiotensin II. Angiotensin II has multiple effects. In addition to contributing to vasoconstriction, angiotensin II also stimulates thirst, promoting the addition of volume to the blood. Angiotensin II also drives the secretion of aldosterone (from the adrenal cortex) and antidiuretic hormone (from the hypothalamus / posterior pituitary), which, among their numerous effects, synergistically increase the retention of salt and water in the kidneys, thereby preventing the loss of current blood volume.
Aldosterone’s primary role is to increase serum sodium levels. In part, this is accomplished by increasing the retention of sodium by the kidneys by driving translocation and activity of sodium pumps and channels. Antidiuretic hormone’s primary peripheral role is to increase the retention of water by the kidneys. This is accomplished by driving the recruitment of aquaporins, which enable the passive flow of water across membranes along osmotic gradients.
Long-Term Responses to Decreased Blood Pressure/Volume
The long-term endocrine responses to decreased blood pressure/volume involve increasing the number of red blood cells. The kidneys release erythropoietin, which signals the production of red blood cells in bone marrow and also promotes the survival of existing red blood cells. Furthermore, erythropoietin induces the release of erythroferrone by bone marrow. The creation of new red blood cells requires the availability of iron, which is necessary for hemoglobin to bind oxygen and carbon dioxide. Erythroferrone from bone marrow increases iron availability by inhibiting the liver’s transcription of hepcidin, which otherwise reduces iron availability. Iron availability is also important in the mid-term responses to decreased blood volume because iron is necessary for the production of aldosterone (more on this shortly).
Before an in-depth examination of the mid-term endocrine responses to decreased blood pressure/volume, it is important to understand basic renal physiology because the mid-term responses to decreased blood pressure/volume are largely accomplished through changes in renal filtration.
 Renal Physiology
Renal Physiology
The primary role of the renal system is to filter blood. This is accomplished by the primary organs of the renal system, the kidneys. One kidney is on the right side of the posterior abdominal cavity, and the other kidney is on the left side. The right kidney is usually slightly smaller and lower in the abdominal cavity than the left kidney because it is displaced by the liver.
Each kidney is composed of a renal cortex surrounding the renal medulla. The renal medulla is divided into renal pyramids. The kidneys filter blood by nonselectively dumping all blood, and then selectively re-uptaking only the desired components of blood. In fact, the kidneys are so efficient at filtering blood that only half of one kidney is necessary to efficiently filter blood. Blood is supplied to the kidneys by renal arteries, which arise from the abdominal aorta. At any given time, up to a third of total cardiac output may pass through the kidneys for filtration.
The functional units of the kidneys capable of this filtration process are called nephrons, of which there are about one-million per kidney. Renal arteries branch to supply blood to various nephrons. TOnce blood is filtered into Bowman’s capsule, it is called filtrate.
hese afferent blood vessels carry blood into a specialized capillary bed within each nephron, called the glomerulus. At this point, blood is nonselectively dumped into at a region called Bowman’s capsule, in the renal cortex. Podocytes are fenestrated cells that line the glomerulus, and allow all components of blood to be dumped into Bowman’s capsule except for cells, platelets, and certain large proteins. While some water is dumped into bowman’s capsule, water also remains in the blood due to the plasma oncotic pressure, a form of osmotic pressure created by large proteins (primarily albumin, from the liver), which pull water into the circulatory system and prevent water from leaking out of the circulatory system.
After being dumped into Bowman’s capsule, the filtrate then travels through the proximal convoluted tubule of the nephron. In the proximal convoluted tubule, amino acids, nucleic acids, nutrients, vitamins, and ions, are actively pumped out of the nephron, and resorbed back into the blood. The proximal tubule then begins to thin as it passes out of the renal cortex and into the renal medulla. This is the beginning of the descending limb of the loop of Henle. The deeper into the renal medulla, the higher the solute concentration. This permits the osmosis of water through aquaporins (passive water transporters) out of the nephron for reabsorption.
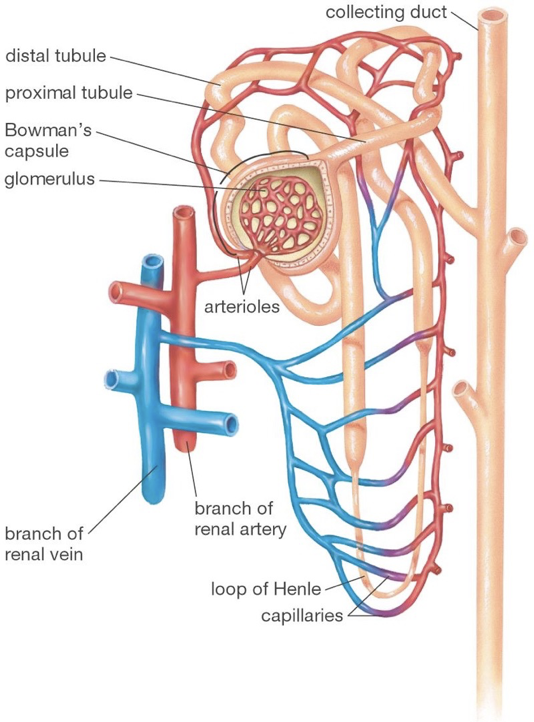
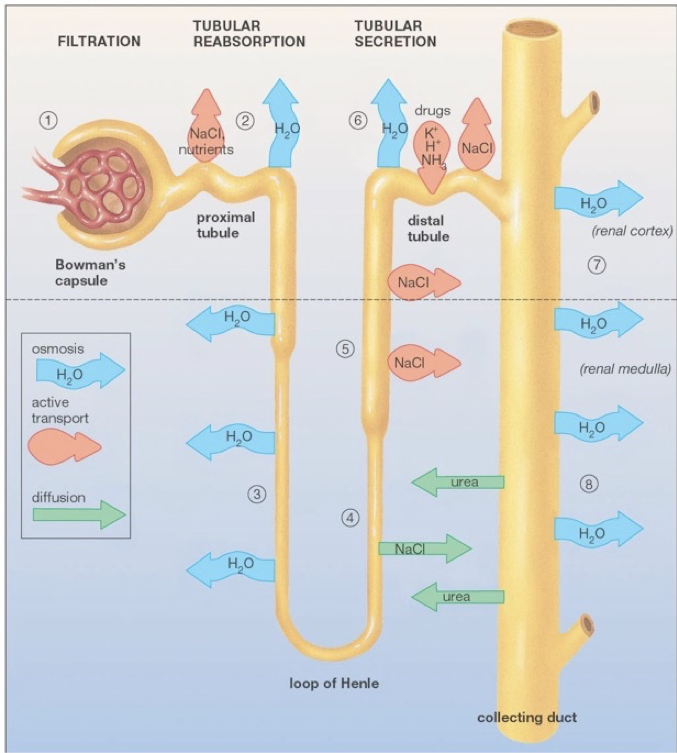 The descending limb of the loop of Henle is only permeable to water. As water travels further down the loop of Henle, it will always be attracted to the increasingly solute-rich environment of the renal medulla, despite the increasing concentration of solutes in the nephron. The loop of Henle rounds, and ascends (the ascending limb) back up the renal medulla toward the renal cortex. As the highly concentrated contents of the nephron (soon to be urine) ascends though the thin ascending limb, the external environment becomes less rich in solutes, allowing some ions (e.g., sodium and chloride) to diffuse down their concentration gradients out of the nephron through passive ion channels. There are no aquaporins on the ascending limb. The ascending limb then thickens, becoming the thick ascending limb, at which point ions are actively transported out of the nephron. The point at which aquaporins first appear again is the beginning of the distal convoluted tubule. The distal convoluted tubule is one of the primary points at which the kidneys are regulated by hormones and by drugs. In addition to water’s osmosis out of the distal convoluted tubule, sodium and chloride ions are actively pumped out of the filtrate. Furthermore, potassium, hydrogen, and ammonia (among other things), are actively pumped into the distal convoluted tubule. The distal tubule then connects with a collecting duct, which descends, again, though the renal medulla. More aquaporins line the collecting duct, and allow osmosis as the external environment, again, becomes increasingly rich in solutes. Collecting ducts connect to the renal pelvis, which funnels urine into a ureter that carries urine to the bladder for excretion.
The descending limb of the loop of Henle is only permeable to water. As water travels further down the loop of Henle, it will always be attracted to the increasingly solute-rich environment of the renal medulla, despite the increasing concentration of solutes in the nephron. The loop of Henle rounds, and ascends (the ascending limb) back up the renal medulla toward the renal cortex. As the highly concentrated contents of the nephron (soon to be urine) ascends though the thin ascending limb, the external environment becomes less rich in solutes, allowing some ions (e.g., sodium and chloride) to diffuse down their concentration gradients out of the nephron through passive ion channels. There are no aquaporins on the ascending limb. The ascending limb then thickens, becoming the thick ascending limb, at which point ions are actively transported out of the nephron. The point at which aquaporins first appear again is the beginning of the distal convoluted tubule. The distal convoluted tubule is one of the primary points at which the kidneys are regulated by hormones and by drugs. In addition to water’s osmosis out of the distal convoluted tubule, sodium and chloride ions are actively pumped out of the filtrate. Furthermore, potassium, hydrogen, and ammonia (among other things), are actively pumped into the distal convoluted tubule. The distal tubule then connects with a collecting duct, which descends, again, though the renal medulla. More aquaporins line the collecting duct, and allow osmosis as the external environment, again, becomes increasingly rich in solutes. Collecting ducts connect to the renal pelvis, which funnels urine into a ureter that carries urine to the bladder for excretion.
The flow of blood in peritubular capillaries runs counter to the flow of urine in the nephron throughout this process, particularly in the loop of Henle; blood descends along the ascending limb of the loop of Henle and ascends along the descending limb of the loop of Henle. This allows countercurrent exchange to occur. In the descending limb of the loop of Henle, thereIf calcium excretion is particularly high (hypercalciuria) and concentrated, calcium can form kidney stones in the renal pelvis.
is countercurrent exchange of water, and in the ascending limb, there is countercurrent exchange of sodium and chloride. In the descending limb of the loop of Henle, the filtrate becomes increasingly concentrated as water moves into the more highly-concentrated interstitial fluid. At the end of the descending limb, before entering the ascending limb, the filtrate is most concentrated. In the ascending limb, the filtrate becomes increasingly dilute as sodium and chloride diffuse into the interstitial fluid along their concentration gradients. Before flowing down the ascending limb of the loop of Henle, blood in peritubular capillaries has a lower concentration of sodium and chloride than the interstitial space. As a result, sodium and chloride diffuse from the interstitial space into peritubular capillaries along their concentration gradients. As blood descends into the renal medulla, it becomes increasingly concentrated, but due to the increasing concentration of solutes in the interstitial space, the sodium and chloride continue to diffuse into the peritubular capillaries along their concentration gradients. As blood in peritubular capillaries begins to flow up the descending limb, the blood is more concentrated than the interstitial fluid. As a result, water enters the peritubular capillaries along its osmotic gradient. While this causes the blood to become increasingly dilute as the peritubular capillaries ascend, the blood is always slightly more concentrated than the surrounding interstitial fluid, maintaining an osmotic gradient that attracts water from the interstitial fluid.
Mid-Term Responses to Decreased Blood Pressure/Volume (In-Depth)
Mid-term responses to decreased blood pressure/volume are accomplished through the RAAS signaling pathway.
Renin Converts Angiotensinogen to Angiotensin I
The renin-angiotensin-aldosterone (and antidiuretic hormone) signaling pathway begins with the production and secretion of renin by the kidneys. Renin is produced from prorenin by juxtaglomerular cells.
Within each nephron, juxtaglomerular cells surround the afferent arterioles that deliver blood to each glomerulus. Juxtaglomerular cells constitutively produce and release prorenin. Other tissues also release prorenin, including the adrenal glands and gonads (i.e., female ovaries and male testes). In fact, circulating levels of prorenin are about ten times higher than those of renin. This provides sufficient prorenin for conversion to renin.
Juxtaglomerular cells produce and release renin in response to multiple stimuli, including falling blood pressure, decreased pulse amplitude (detected by baroreceptors), excessive urine production, or in response to sympathetic activity (via β1 adrenergic receptors). While juxtaglomerular cells have baroreceptors that directly measure the mechanical stretch of the afferent arteriole to provide an indication of blood pressure/volume, information about urine flow is provided to juxtaglomerular cells though paracrine signals (e.g., prostaglandin E2) released by macula densa cells.
While renin is commonly referred to as a hormone, renin is functionally an enzyme; renin is sometimes called angiotensinogenase. Once released into circulation, renin converts the blood borne protein angiotensinogen (a pre-pro-hormone produced by the liver) to angiotensin I (AI).
Macula densa cells are located on the distal convoluted tubules of nephrons, adjacent to the juxtaglomerular apparatus. This enables efficient paracrine signaling from macula densa cells to juxtaglomerular cells. While there are only about 20 macula densa cells per nephron, they are well attuned to sensing variations in the fluid microenvironment of the distal convoluted tubule (e.g., the concentrations of salts and metabolites, and the flow rate of the filtrate).
Macula densa cells sense tubular concentrations of sodium-chloride via the sodium-potassium-chloride cotransporter 2 (NKCC2), which allows an influx of sodium, potassium, and chloride using the concentration gradient of sodium (more on the NKCC2 later).
When there is a low influx of sodium-chloride through the NKCC2, there is a lower concentration of sodium-chloride within macula densa cells. In response to decreased intracellular concentrations of sodium-chloride, there is increased activation of cyclooxygenase-2 and microsomal prostaglandin E synthase. Increased activity in these enzymBecause ACE can limit the production of angiotensin II, and can thereby prevent consequent increases in blood pressure/volume, ACE inhibitors are an effective treatment of high blood pressure. However, ACE inhibitors only function to a certain limit; the effects of ACE inhibitors can be overwhelmed by high release and activity of renin. This may be problematic because of the nature of the RAAS signaling pathway. For example, when an individual takes an ACE inhibitor, it prevents the production of angiotensin II. This removes angiotensin II-mediated feedback on renin release (via the AT1 receptor). As a consequence, renin levels will rise, and overwhelm the effects of the ACE inhibitor, potentially leading to high levels of angiotensin II and consequent high blood pressure/volume. To prevent this risk, diuretics may be administered with ACE inhibitors to reduce water retention in the kidneys, and lower blood volume.
es increases the production of prostaglandin E2. Prostaglandin E2 serves as a paracrine signal to juxtaglomerular cells. When prostaglandin E2 from macula densa cells binds to the EP4 receptor on juxtaglomerular cells, it induces the production and release of renin. Thus, when there are low sodium-chloride concentrations in the distal convoluted tubule, macula densa cells signal the production and release of renin by juxtaglomerular cells.
Angiotensin Converting Enzyme Converts Angiotensin I to Angiotensin II
Angiotensin I is converted to angiotensin II by angiotensin converting enzyme (ACE), which is found in the lungs. ACE only operates when the lungs permit it. The activity of ACE depends on the amount of fluid passing through pulmonary tissues. If blood pressure is high, blood in the lungs loses fluid in the pleural cavity as it leaks out of weak pulmonary capillaries. This fluid loss in the pleural cavity can lead to a pulmonary edema. The lungs minimize this risk by preventing the conversion of angiotensin I to angiotensin II by ACE.
Angiotensin II
Angiotensin II (AII) has multiple effects. Logically, angio–tensin II causes blood vessel tension (i.e., angiotensin II promotes vasoconstriction). It also stimulates the secretion of aldosterone by the adrenal cortex, as well as the release of antidiuretic hormone from the posterior pituitary (i.e., by the hypothalamus). Angiotensin II even stimulates thirst at the level of the hypothalamus to increase consumption of fluids that will contribute to raising blood volume. However, one especially important function of angiotensin II is to exert negative feedback upon the production of renin by juxtaglomerular cells. When angiotensin II binds to AT1 receptors on juxtaglomerular cells, it inhibits the production of renin.
Aldosterone
Aldosterone (ALDO) is a mineral corticoid (steroid hormone) produced in a distinct region of the adrenal cortex called the zona glomerulosa. The zona glomerulosa is a thin, 10-20 cell-thick layer just below the surface of the adrenal cortex. All aldosterone synthesis takes place in the mitochondria of cells in this discrete region. Mineral corticoids (such as aldosterone), glucocorticoids, androgens, estrogens, and progestogens are all derived from cholesterol through multi-step synthesis involving multiple enzymes. Thus, cholesterol is the precursor to aldosterone. Aldosterone synthase [ADX; also called P450 (11β2)] is the enzyme that synthesizes aldosterone from the immediate precursor corticosterone. The activity of aldosterone synthase is dependent on the presence of adrenal ferredoxin (also called adrenodoxin), which serves as a cofactor by transporting iron into mitochondria to aid in electron transport. This also means that adequate iron availability is necessary for the production of aldosterone. Enzymes in the adrenal cortex responsible for the synthesis of cholesterol-based hormones have reducing and producing capabilities, each specific to a particular moiety. Aldosterone synthase is only present in the zona glomerulosa, and is imperative for survival. Without it, death is inevitable. The adrenal cortex has a limited supply of cholesterol, and a limited number of enzymes, both of which can serve as rate-limiting factors. Due to the reliance on adrenal ferredoxin (and therefore iron) as a cofactor in ALDO production, hypoaldosteronism can result from a mutation in the ferredoxin gene or from conditions that result in insufficient levels of serum iron.
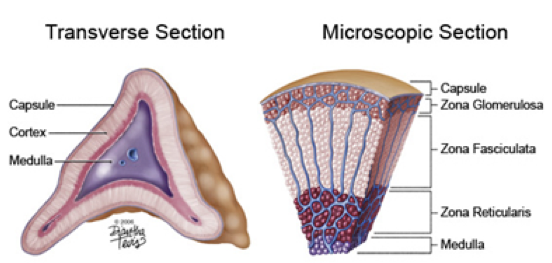
The adrenal gland is divided into the adrenal medulla and adrenal cortex. In the center of the adrenal gland, the adrenal medulla is composed of axon terminals of sympathetic neurons that produce and release catecholamines (primarily epinephrine and norepinephrine). Surrounding the adrenal medulla is the adrenal cortex, which is divided into three layers. Immediately surrounding the adrenal medulla is the zona reticularis of the adrenal cortex, which produces and secretes adrenal androgens. Surrounding the zona reticulata is the zona fasciculata of the adrenal cortex, which produces and secretes glucocorticoids (e.g., cortisol). Finally, the outermost layer of the adrenal cortex is the zona glomerulosa, which produces and secretes mineral corticoids (e.g., aldosterone).
Under normal conditions, there is a basal release rate of aldosterone, so signals enhance or inhibit the basal rate of release. In other words, aldosterone will normally be released in the absence of regulatory hormones.
Angiotensin II and extracellular potassium concentration are the two major driving forces of aldosterone release. Despite aldosterone’s role in increasing and maintaining circulating sodium levels, low sodium is less of a stimulus than high potassium. High potassium levels are a stronger indication of physiological vulnerability; there must be a balance between sodium and potassium levels for normal functioning. Potassium usually antagonizes the effects of sodium, so their relative concentrations are important. High potassium levels have similar effects as low sodium levels.
Cells of the zona glomerulosa have innate electrical excitability. When these cells are depolarized sufficiently, they open voltage-gated calcium channels. The consequent influx of calcium initiates the release of aldosterone. Cells of the zona glomerulosa constitutively express inward-rectifying potassium channels.
Potassium is largely responsible for maintaining a hyperpolarized membrane potential in most cells (including cells of the zona glomerulosa). Inward-rectifying potassium channels (Kir) on the cell membrane remain open, permitting the movement of potassium along its concentration and electrical gradients. Potassium is more concentrated inside cells than outside, so the concentration gradient is always driving potassium out of cells (this intracellular concentration is maintained by sodium-potassium-ATPase pumps). When the membrane is hyperpolarized to a certain point (i.e., with the interior more negatively charged than the exterior), potassium ions will have no net movement because they are equally attracted to the negative cell interior and repelled by the high concentration of intracellular potassium. This is the equilibrium potential of potassium. When the membrane is depolarized from this negative membrane potential, the electrical attraction to the negative interior is reduced. Thus, the concentration gradient will drive potassium ions out of the cell, and the efflux of positive charges will return the membrane potential towards the equilibrium potential of potassium. When the membrane is hyperpolarized beyond potassium’s equilibrium potential, potassium ions will be more attracted to the negatively charged interior than they are repelled by the high concentration of potassium, and will enter the cell. The influx of positively-charged potassium ions returns the membrane potential to the initial level of hyperpolarization. However, potassium is not the only ion permeable at rest. Other ions permeable at rest (e.g., sodium) are constantly driving the membrane towards less hyperpolarized membrane potentials (e.g., sodium is always flowing into the cell to some extent, which depolarizes the membrane potential). Because potassium is highly permeable relative to other ions at rest, a constant potassium efflux is able to counteract these depolarizing influences and maintain a somewhat fixed membrane potential. In other words, there is a balance between depolarizing ion flow and the hyperpolarizing potassium efflux. However, this is a delicate balance, and can only be maintained to the extent that potassium is permeable. If signals increase the permeability of ions that depolarize the membrane, they may overcome the ability of potassium efflux to hyperpolarize the membrane, resulting in a net depolarization. Likewise, if signals block the efflux of potassium to some extent, the balance will be disrupted to favor the ever-present depolarizing influences, which also results in a net depolarization of the membrane. If depolarization is sufficient to activate voltage-gated calcium channels, the consequent influx of calcium will initiate the secretion of hormones.
Factors that promote aldosterone secretion tend to do so by reducing potassium efflux, which enables depolarizing ion flow to raise the membrane potential to the point at which voltage-gated calcium channels open.
Angiotensin II binding to AT1 receptors on zona glomerulosa cells initiates a signaling cascade that closes potassium channels. This results in a shift in the balance between the depolarizing ion flow and hyperpolarizing potassium efflux, favoring the flow of ions that depolarize the membrane. The depolarization opens voltage-gated calcium channels, and the consequent calcium influx initiates aldosterone secretion.
Without changing potassium permeability, high serum potassium changes the concentration gradient governing the movement of potassium across the membrane of zona glomerulosa cells. Remember that the internal potassium concentration remains functionally constant due to the efficiency of the sodium-potassium-ATPase pump. However, by raising the extracellular concentration of potassium, the concentration gradient driving potassium efflux is reduced (in other words, the equilibrium potential for potassium is less polar when there is a high extracellular concentration of potassium). As a result of this reduction in potassium efflux, the membrane will depolarize, voltage-gated calcium channels will open, and aldosterone will be secreted.
However, angiotensin II and high serum potassium are not the only aldosterone release stimuli. Angiotensin III, a breakdown product of angiotensin II, also simulates aldosterone release with equal efficacy to angiotensin II. Furthermore, adrenal innervation allows the sympathetic nervous system to initiate aldosterone release when baroreceptors in the carotid artery detect low blood pressure, indicating a shortage of blood flow to the brain. Finally, high levels of stress can increase the rate of aldosterone release.
Aldosterone secretion is affected by indirect inputs from the hypothalamus, as part of the Hypothalamus-Pituitary-Adrenal Gland Axis (HPA axis). The HPA axis is similar to the HPT axis, which governs thyroid hormone synthesis and release. Like the HPT axis, the HPA axis begins with the hypothalamic release of a releasing hormone into closed circulation with the anterior pituitary. The releasing hormone signals the anterior pituitary to release a tropic hormone into open circulation. The tropic hormone then effects target organs’ hormone secretion. In the HPA axis, the hypothalamus releases corticotropin-releasing hormone, which signals the anterior pituitary to release adrenocorticotropic releasing hormone (ACTH, also called adrenocorticotropin) into open cInsulin-releasing β cells of the pancreas behave in a similar fashion to aldosterone-releasing cells. In pancreatic β cells, potassium is also primarily responsible for establishing a resting membrane potential. In β cells, the closure of ATP-sensitive potassium channels causes the membrane to depolarize, opening voltage-gated calcium channels that allow for an influx of calcium, which drives insulin secretion.
irculation. ACTH stimulates the production of corticosteroids from the adrenal cortex. ACTH has a large impact on the production and secretion of glucocorticoids in the zona fasciculata. However, ACTH only has a minor impact on the production (but not release) of aldosterone by stimulating production of the aldosterone precursor, deoxycorticosterone (deoxycorticosterone is the precursor to corticosterone, which is converted to aldosterone by aldosterone synthase). There are very few ACTH receptors on the zona glomerulosa, and they are quickly saturated, causing ACTH-driven aldosterone synthesis to rapidly reach its peak effects. Thus, ACTH effects on circulating levels of aldosterone are minimal, especially in contrast to the proportional effects of ACTH on circulating levels of glucocorticoids (e.g., cortisol).
Once released, aldosterone is free to bind to mineral corticoid receptors (MRs). While some aldosterone receptors are expressed on membrane surfaces, aldosterone generally exerts its effects through cytoplasmic receptors that alter transcription. Activation of aldosterone receptors has multiple effects, including the stimulation of sodium retention by the kidneys, inhibition of potassium retention by the kidneys, and the stimulation of sodium uptake by the colon.
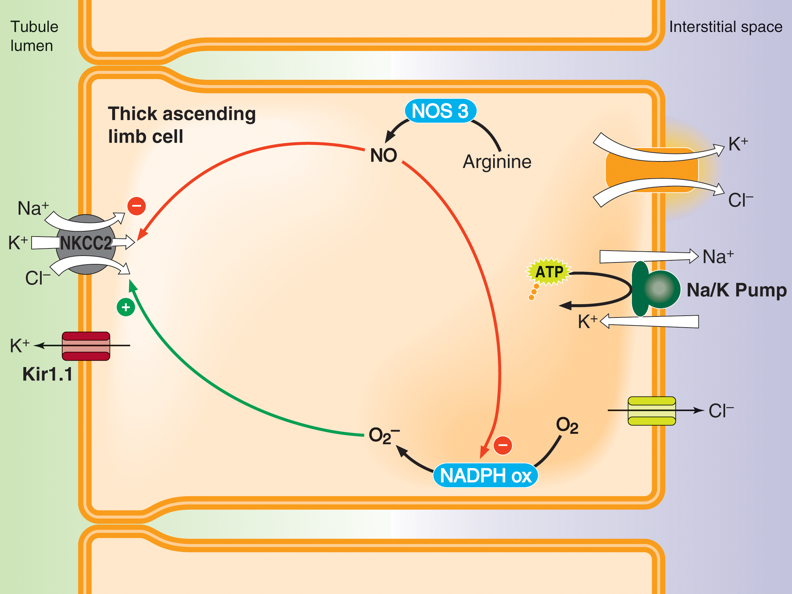 In the kidneys, aldosterone increases the reuptake of sodium in the thick ascending limb of the loop of Henle, in the distal convoluted tubule, and in the collecting duct. Firstly, aldosterone upregulates amiloride-sensitive sodium channels, which permit the passive movement of sodium along its concentration gradient. Aldosterone also upregulates the sodium-potassium-chloride cotransporter 2 (NKCC2) in the thick ascending limb and distal convoluted tubule both by increasing transcription of NKCC2, and also by increasing the activity of NKCC2. NKCC2 is expressed on tubule cell membranes exposed to the lumen of the nephron (i.e., NKCC2 is expressed on the apical membranes of epithelial tubule cells). Using the concentration gradient of sodium, NKCC2 transports sodium, potassium, and chloride from the filtrate into these tubule cells.
In the kidneys, aldosterone increases the reuptake of sodium in the thick ascending limb of the loop of Henle, in the distal convoluted tubule, and in the collecting duct. Firstly, aldosterone upregulates amiloride-sensitive sodium channels, which permit the passive movement of sodium along its concentration gradient. Aldosterone also upregulates the sodium-potassium-chloride cotransporter 2 (NKCC2) in the thick ascending limb and distal convoluted tubule both by increasing transcription of NKCC2, and also by increasing the activity of NKCC2. NKCC2 is expressed on tubule cell membranes exposed to the lumen of the nephron (i.e., NKCC2 is expressed on the apical membranes of epithelial tubule cells). Using the concentration gradient of sodium, NKCC2 transports sodium, potassium, and chloride from the filtrate into these tubule cells.
Aldosterone also increases the number and activity of sodium-potassium-ATPase pumps in tubule cells. These sodium-potassium-ATPase pumps exchange intracellular sodium for extracellular potassium. Thus, sodium ions that enter the tubule cells through the NKCC2 are pumped into the interstitial fluid, where they can be absorbed by peritubular capillaries. Chloride that entered the tubule cells via the NKCC2 is also absorbed by peritubular capillaries after either passively flowing into the interstitial space through chloride channels or being cotransported with potassium into the interstitial space using the high intracellular concentration of potassium.
Aldosterone also increases the expression of potassium channels on the apical membrane of epithelial tubule cells. These potassium channels allow the accumulating intracellular potassium within these tubule cells to flow iPotassium pumps in the colon actively pump potassium from the colon into the bloodstream, creating an osmotic gradient that enables the absorption of water. Cholera in intestinal cells increases cAMP production, which activates PKA. PKA induces the activity of CFTR chloride pumps. The active pumping of chloride into the intestines creates an osmotic drive that pulls water and salts out of the body, resulting in an inability to efficiently absorb water and consequent dehydration.
nto the filtrate along its concentration gradient.
Thus, at the level of the nephron, aldosterone increases the reabsorption of sodium, while decreasing the reabsorption of potassium. These channels and transporters are the targets of numerous drugs, such as heart and reproductive medications. The sodium-potassium-ATPase pump, though, is a target of some poisons, such as glycosides and digoxin.
Aldosterone also enhances the amount of sodium absorbed by the body in the first place by increasing sodium uptake by the colon. In the colon, almost everything is waste material and bacteria, but it is also the site of water absorption. Aldosterone upregulates amiloride-sensitive sodium channels in the intestinal tract, which permits the absorption of sodium as it flows along its concentration gradient. Furthermore, aldosterone also stimulates sodium potassium exchange, resulting in increased sodium absorption, and the excretion of potassium. This promotion of sodium-potassium exchange is not limited to the intestinal tract. Aldosterone also promotes sodium-potassium exchange by the salivary glands and by sweat glands.
When there is deficient aldosterone production, there is potential for hyperkalemia, a decrease in muscle control and strength, a decrease in blood pressure, and neurological effects. These symptoms of hypoaldosteronism are also present in hypothyroidism. Hyperaldosteronism, on the other hand, is less frequent. Among other causes, hyperaldosteronism may result when a maternal effect gene (inherited from the mother) causes mitochondria in the zona glomerulosa to divide.
Antidiuretic Hormone
Antidiuretic hormone (ADH, also called vasopressin) is produced in magnocellular neurons of the paraventricular nucleus of the hypothalamus and in the supraoptic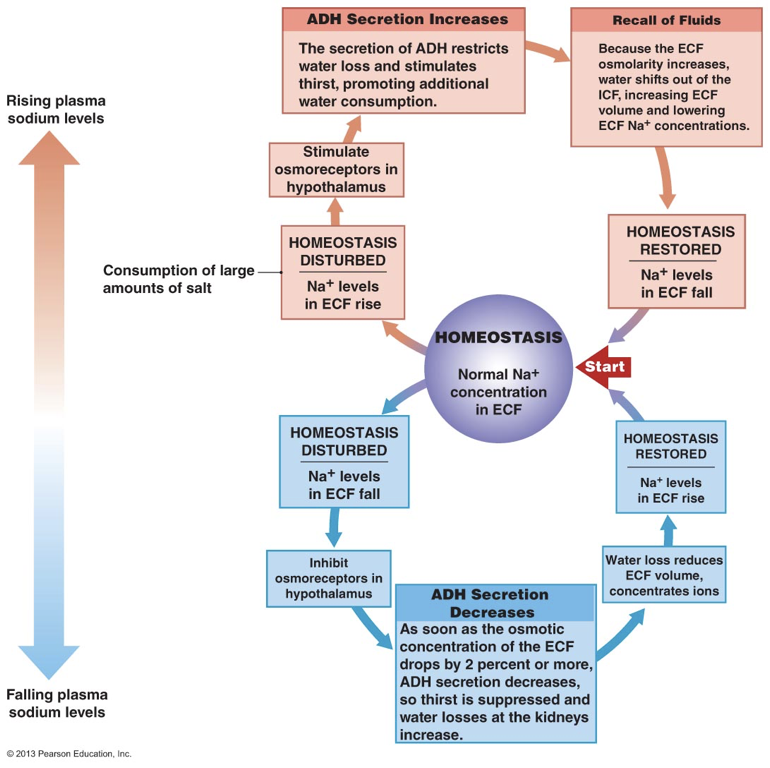 nucleus of the hypothalamus. After being produced in these nuclei, but without ever leaving the magnocellular neurons, ADH is transported along magnocellular axons, through the infundibulum, to ax
nucleus of the hypothalamus. After being produced in these nuclei, but without ever leaving the magnocellular neurons, ADH is transported along magnocellular axons, through the infundibulum, to axThe glucocorticoid cortisol is also capable of binding to mineral corticoid receptors. However, 11 β2-hydroxysteroid dehydrogenase (11 β2-HSD) is co-localized with mineral corticoid receptors. 11 β2-HSD converts cortisol to cortisone, which cannot effectively activate the receptor. This prevents mineral corticoid receptor activation by glucocorticoids, which would be problematic in cases where glucocorticoid levels are high even when aldosterone levels are low (i.e., because there is no need to increase serum sodium).
on terminals in the posterior pituitary, from which ADH is stored until signals initiate its release into open circulation.
Angiotensin II drives the secretion of ADH from the posterior pituitary, but secretion is also governed by deviations in homeostatic parameters. For example, ADH is secreted when the hypothalamus detects high plasma osmolarity. High plasma sodium concentrations are accompanied by high sodium concentrations in the extracellular fluid in the hypothalamus. High extracellular sodium is detected by hypothalamic osmoreceptors, which signal for the secretion of ADH. Since ADH increases water volume, ADH is able to dilute high concentrations of sodium. ADH is also secreted when the hypothalamus receives signals of decreased arterial blood volume, even in the presence of low plasma osmolarity (e.g., during a period of hyponatremia). Importantly, ADH is not always released. In other words, there is not necessarily a basal rate of ADH release. This is in stark contrast to aldosterone.
After being released into open circulation, ADH has multiple effects. Firstly, one of the central effects of ADH is to promote thirst, increasing consumption of water, which raises the volume of water in blood. ADH also increases water reabsorption in the kidneys, minimizing the loss of water in urine. ADH stimulates transcription and translocation of aquaporins to the apical membranes of epithelial cells lining the distal convoluted tubule and collecting duct. This enhances the ability of water to flow along its osmotic gradient out of the filtrate and into the interstitial space, from which it is absorbed by peritubular capillaries. This effect on aquaporin transcription and translocation is a short, 15-20 minute long effect because of the rate at which proteins (including aquaporins) are recycled.
ADH also increases the permeability of the collecting duct to urea, which contributes to the osmotic gradient drawing water into the interstitial space. Lastly, ADH enhances sodium reabsorption in the ascending limb of the loop of Henle by driving NKCC2 trafficking, and by driviIn the context of renal physiology, antidiuretic hormone is regularly used due to its descriptive name. However, when discussing the hormone in other contexts, such as its role as a neurotransmitter in neurophysiology, vasopressin is more commonly used. Both terms refer to the same hormone. When reading the abbreviation of antidiuretic hormone, ADH, be aware that the abbreviation is often used for other purposes. For example, ADH is also the abbreviation for alcohol dehydrogenase.
ng phosphorylation of regions of NKCC2 that increase its activity. By increasing sodium reabsorption, ADH enhances to the osmotic gradient that enables water reabsorption.
Alcohol inhibits the secretion of ADH by the posterior pituitary (hypothalamus). Alcohol directly affects the brain, and indirectly affects the kidneys’ ability to retain water by removing the influence of ADH. In rats, alcohol has been shown to block voltage-gated calcium channels that would otherwise initiate the release of ADH from hypothalamic axon terminals in the posterior pituitary. This explains why drinking alcohol leads to the production of large volumes of dilute urine and consequent dehydration.
Despite both of their releases being driven by angiotensin II, ADH and aldosterone operate independently. They have different receptors, and largely act on different targets. Despite operating independently, the two hormones often operate synergistically. For example, by promoting the retention of sodium in the kidneys, aldosterone contributes to the osmotic gradient that enables ADH-mediated increases in water retention. There are situations in which the balance between ADH and aldosterone production is skewed, such that one is produced more or less than the other. For example, in the case of hypernatremia, the high osmolarity in extracellular fluids will drive the release of ADH without promoting the release of aldosterone.
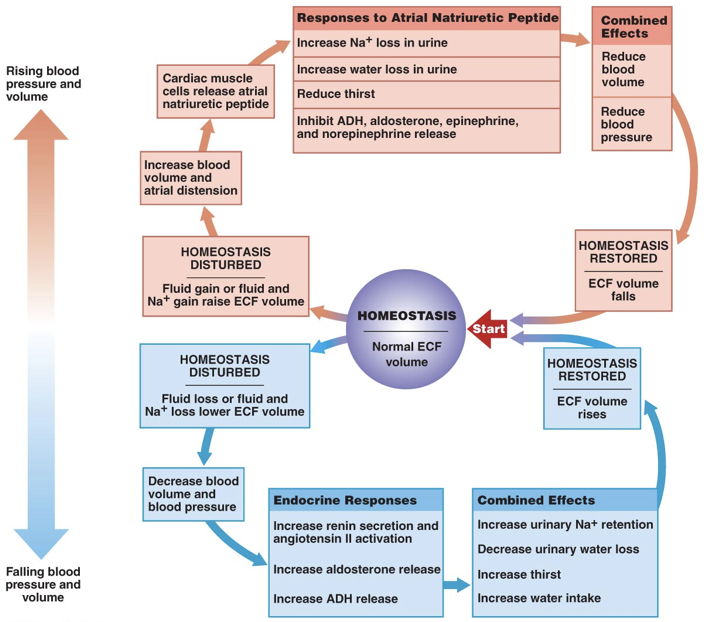
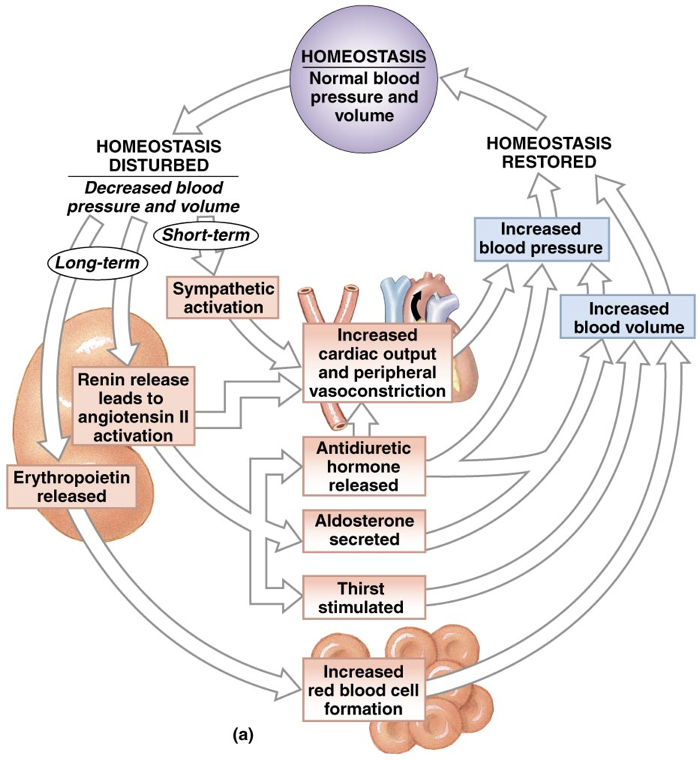
The actions of aldosterone and ADH serve to raise blood sodium and consequently blood pressure. Atrial natriuretic peptide (ANP), on the other hand, serves to lower blood pressure. ANP has effects almost exactly opposite those of aldosterone. ANP is released by the right atrium of the heart when it is over-stretched, or when baroreceptors in the aorta and carotid artery signal excessive blood pressure. Increased blood pressure initiates the release of ANP. ANP decreases sodium levels in the blood by inhibiting the release of aldosterone and by inhibiting the action of ADH in the kidneys. This prevents the retention of sodium and water, leading to lower blood volume and lower blood pressure. Furthermore, ANP inhibits the release of epinephrine and norepinephrine, minimizing their ability to raise blood pressure through vasoconstriction. Brain natriuretic peptide (BNP), despite its misleading name, is primarily released by the ventricles in response to stretching. BNP has effects on blood volume similar to ANP. High levels of BNP are associated with a heightened risk of heart failure.
Long Term Responses to Decreased Blood Pressure/Volume (In-Depth)
In addition to its role in retaining or excreting various quantities of water and sodium in accordance with the activity of aldosterone and ADH, the kidneys also have endocrine activity themselves. As mentioned, the kidneys release the enzyme renin. If, in response to decreased blood pressure/volume, the short-term and mid-term responses fail to sufficiently enhance oxygen carrying capacity and the ability to deliver oxygen to tissues where it is needed (measured by the oxygen availability at the kidneys), the kidneys will begin a multi-week process of increasing the number of red blood cells (RBCs), and the longevity of red blood cells. This is achieved through the release of the hormone erythropoietin (EPO), which is both a growth factor (increases blood tissue growth), and a cytokine (increases cell differentiation). In addition to EPO, the kidneys can also release thrombopoietin (TPO) to stimulate the production of platelets (thrombocytes), and leukopoietin (LPO) to stimulate the production of white blood cells (leukocytes).
EPO stimulates the production of more red blood cells and increases the survival red blood cells. This contributes to raising blood volume and also increases oxygen-carrying capacity. Of the roughly 37.2 trillion cells in the body, about 20-30 trillion are red blood cells. This means that red blood cells constitute about 70% of all cells within the body.
Prenatal EPO is produced in the liver. In adults, though, EPO is predominantly produced in the kidneys (specifically in peritubular interstitial fibroblasts). However, the liver retains its ability to produce EPO in response to moderate or severe hypoxia (i.e., low oxygen). Furthermore, EPO can also be produced in the brain (by both neurons and glia), lungs, heart, bone marrow, spleen, hair follicles, osteoblasts, and the reproductive tract.
Endurance athletes (e.g., cyclists) are always trying to find ways to increase circulating EPO because of its ability to increase aerobic capacity. One common method of increasing aerobic capacity is to train at high altitudes (i.e., in lower atmospheric oxygen concentrations), which increases circulating levels of endogenously-synthesized EPO. However, certain athletes are involved in “blood doping,” in which EPO is injected. Blood doping has been shown to increase performance by up to 40%. EPO is a long protein that is modified in numerous locations by sugars; it is heavily glycosylated. In fact, 40% of the molecular weight of EPO is derived from its carbohydrate component. When EPO is synthesized exogenously, these modifications are absent, which enables the detection of blood doping in athletes that inject exogenously-produced EPO. Despite the advantages of blood doping to increase aerobic capacity, increased circulating EPO can lead to blood clots, ischemic episodes (ischemia is a local loss of oxygen supply), pulmonary problems, and kidney damage.
The EPO receptor lacks intrinsic catalytic function. However, upon EPO binding, the EPO receptor associates with tyrosine kinase Janus Kinase 2 (JAK2), which phosphorylates tyrosine residues on the receptor, providing multiple docking sites for signal-transducing proteins. These signaling cascades have numerous effects in different types of cells. One of these signaling cascades begins when signal transducer and activator of transcription 5 (STAT5) binds to one of the tyrosine residues phosphorylated by JAK2. After STAT5 binds to a phosphorylated tyrosine residue, JAK2 phosphorylates a tyrosine residue on STAT5, which causes STAT5 to dissociate. After dissociating, STAT5 can dimerize or tetramize with other STAT proteins, and act as transcription factors. In erythroblasts (red blood cell precursors in bone marrow), EPO activation of a JAK2/STAT5 pathway results in the synthesis and secretion of erythroferrone, which increases iron availability necessary for red blood cell synthesis. EPO receptor activation stimulates red blood cell production (erythropoiesis) and survival and promotes longer lifespans for pluripotent bone marrow cells that are signaled to differentiate into red blood cells. All of this is accomplished through changes in gene expression.
One particularly interesting effect of EPO is that it can increase HbA1c levels. While normally, increases in HbA1c indicate increases in average blood sugar, the increases in HbA1c attributable to EPO are due to the increased lifespan of red blood cells. By increasing the lifespan of red blood cells, there is more time for glucose to bind to hemoglobin, yielding higher levels of glycosylated hemoglobin even when exposed to the same average blood sugar.
Normally, there is a basal release of EPO that compensates for the rate at which red blood cells expire. The life span of a red blood cell is about 4 months. Beyond this basal level of production and release, EPO may also be upregulated.
EPO is transcribed in peritubular interstitial fibroblasts in the kidneys when stimulated by hypoxia inducible factors (HIF), specifically by HIF-2. HIF-2 is a transcription factor that is only able to activate EPO transcription when peritubular interstitial fibroblasts are hypoxic. Hypoxia inducible factors are heterodimers, each composed of an oxygen-sensitive α-subunit and a constitutively expressed β-subunit. There are three α-subunits (HIF-1α, HIF-2α, and HIF-3α). HIF-2 is composed of the HIF-2α subunit and the β-subunit. Under normal oxygen conditions (i.e., normoxia) in peritubular interstitial fibroblasts, 2-oxoglutarate-dependent oxygenases (2OG oxygenases) are able to efficiently hydroxylate proline residues of HIF-2α (and all other α-subunits for that matter). After hydroxylation of these specific proline residues, HIF-2α (and other α-subunits) is targeted and rapidly degraded by VHL-E3 ubiquitin ligase complexes; the von Hippel-Lindau tumor suppressor (VHL) acts as a substrate recognition component (i.e., it recognizes the hydroxylated proline residues) of an E3 ubiquitin ligase. As a consequence, in normal oxygen conditions, HIF-2α is degraded before it can heterodimerize with a β-subunit, preventing its ability to drive transcription of EPO. However, 2OG oxygenases require oxygen to function properly. Under hypoxic conditions, 2OG oxygenase activity is inhibited. This prevents hydroxylation of proline residues on HIF-2α. Without hydroxylation of its proline residues, HIF-2α is not recognizable to VHL, and cannot be degraded by VHL-E3 ubiquitin ligase complexes. Instead, HIF-2α is able to heterodimerize with a β-subunit to form HIF-2, which (along with transcriptional cofactors) is able to drive transcription of EPO. Thus, a hypoxic environment prevents HIF-2α degradation, enabling activation of EPO transcription by the heterodimer HIF-2.
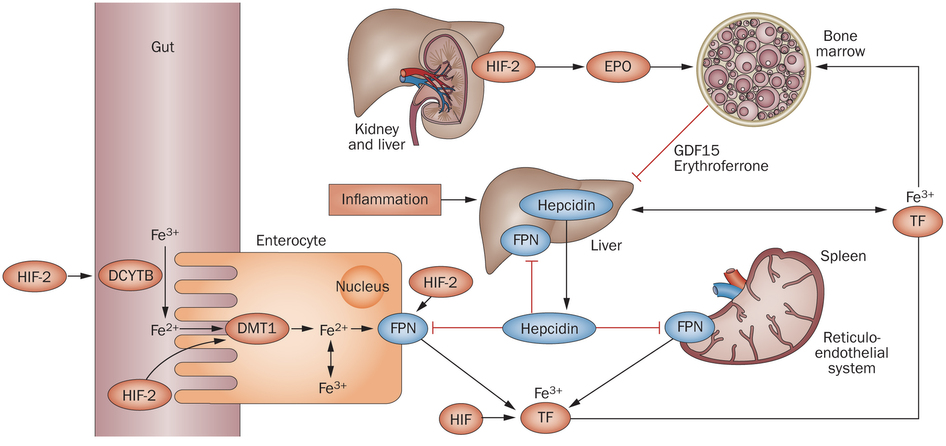
Regulation of iron is also important in the production of red blood cells and in determining cardiac output. In fact, iron is more tightly regulated than most other ions. High levels of iron can cause neurological damage, but even consuming large concentrations of iron does not necessarily increase the circulating iron in the body because regulatory mechanisms permit such fine control. Among many other effects, low levels of iron can lead to hypoaldosteronism due to the use of iron by adrenal ferredoxin, the cofactor of aldosterone synthase.
To produce red blood cells, iron is required. The core of red blood cells is a protein called hemoglobin, which is composed of four subunits, two α-chain subunits, and two β-chain subunits, each of which contains a heme group. Heme groups have iron ions at their center, which allow them to bind oxygen and carbon dioxide. This means that each hemoglobin molecule can bind either four oxygen molecules or four carbon dioxide molecules. In the blood stream, “free” iron is a toxic substance, but it is not toxic when bound to hemoglobin. In other words, hemoglobin is effectively an iron chelator.
Normally, iron is stored in the liver and spleen, where ferroportin transports iron out of cells; ferroportin is the only known iron ion exporter. The spleen is the blood cell salvage pathway, and it holds onto the iron found in hemoglobin. However, if iron levels are high, the liver releases a signal molecule, called hepcidin, into the bloodstream. Hepcidin facilitates the degradation and internalization of ferroportin, thereby preventing iron ions from being released into the blood stream. This not only prevents the release of iron by the liver and spleen, but also prevents the uptake of iron in dietary sources from the intestinal lumen (e.g., after iron is absorbed by enterocytes, there is no way for iron to leave those enterocytes due to blockage of ferroportin, so iron never enters the bloodstream). However, hepcidin is also inhibited during states of hypoxia, permitting increased iron availability for red blood cell production by preventing hepcidin-mediated inhibition of iron export by ferroportin. However, the inhibition of hepcidin in hypoxic conditions is not a direct response to hypoxic conditions like the production of EPO. Instead, hypoxic conditions inhibit hepcidin through a multistep endocrine signaling pathway. In hypoxic conditions, there is increased EPO transcribed in the kidneys, and EPO drives the secretion of erythroferrone by erythroblasts in bone marrow. Erythroferrone is then able to inhibit expression of hepcidin in the liver, resulting in increased activity of ferroportin, which consequently increases serum iron.
pH Regulation
Hemoglobin is composed of four subunits, two α-chains and two β-chains, each of which contains a heme group. Heme groups have iron ions at their center, which allow them to bind oxygen and carbon dioxide. This means that each hemoglobin molecule can bind four oxygen molecules or four carbon dioxide molecules. However, heme groups bind oxygen in an unusual way due to protein cooperativity. Hemes bind oxygen reluctantly; they only bind oxygen in a high oxygen concentration environment. Once one heme group binds an oxygen molecule, the remaining three will immediately bind oxygen as well. Once oxygen is bound, hemoglobin will only release oxygen in areas of very low oxygen concentration. Again, they are reluctant to release oxygen, but as soon as oxygen is released by one heme, the remaining oxygen is released as well. Cooperative proteins operate based on this all or nothing principle. Because of the quaternary structure of hemoglobin, the individual heme groups bind oxygen differently than if they were acting independently. If this cooperative relationship is plotted graphically as the percentage of hemoglobin molecules saturated by oxygen as a function of the concentration of oxygen, the resulting relationship is an S-shaped curve.
Interestingly, the body does not have any oxygen sensors. As oxygen is used, it is converted to carbon dioxide. When carbon dioxide is in water, it forms carbonic acid. Because carbon dioxide is a metabolic byproduct, it is present in regions of high activity (i.e., regions that need oxygen to continue their metabolic processes). Carbonic acid lowers the pH of the blood, making it more acidic.
Proteins behave differently at different pH levels. As described, there is an S-curve graph of the percentage of hemoglobin molecules saturated with oxygen as a function of oxygen concentration. Because hemoglobin is a protein and proteins behave differently at different pH’s, the S-curve relationship is dependent on pH. When the pH becomes more alkaline (basic) as the pH rises, the S-curve will shift up; at every concentration of oxygen, hemoglobin will be more likely to hold onto oxygen, and less likely to release oxygen. However, when the pH lowers, becoming more acidic (e.g., due to concentrated carbonic acid) the S-curve will shift down; at every concentration of oxygen, hemoglobin will be less likely to hold onto oxygen, and more likely to release oxygen. This downward shift in the S-shaped curve is called a Bohr shift. This means that hemoglobin can effectively transport and release oxygen exactly where it is being used due to the low concentration of oxygen and lower pH caused by carbonic acid, both of which are strong indicators that oxygen is needed without requiring oxygen sensors.
Hemoglobin has a similar relationship with carbon dioxide. Hemoglobin will be more likely to bind carbon dioxide in areas of high carbon dioxide concentration and will release carbon dioxide in areas of low carbon dioxide concentration. However, the S-shaped curve representing the percent of hemoglobin bound by carbon dioxide as a function of carbon dioxide concentration will shift up when the pH lowers (more acidic) and shift down when the pH rises (more basic); this is the opposite of how pH affects hemoglobin binding oxygen. This means that hemoglobin will be more likely to bind carbon dioxide in areas of high activity (i.e., where carbon dioxide is produced as a byproduct of metabolism), and release carbon dioxide in the lungs, where there is low carbon dioxide concentration.
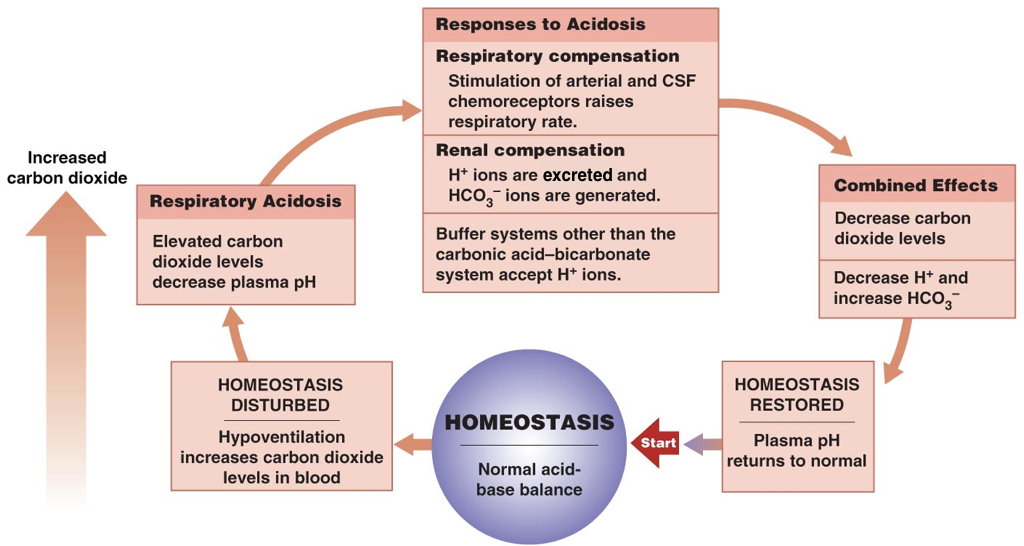
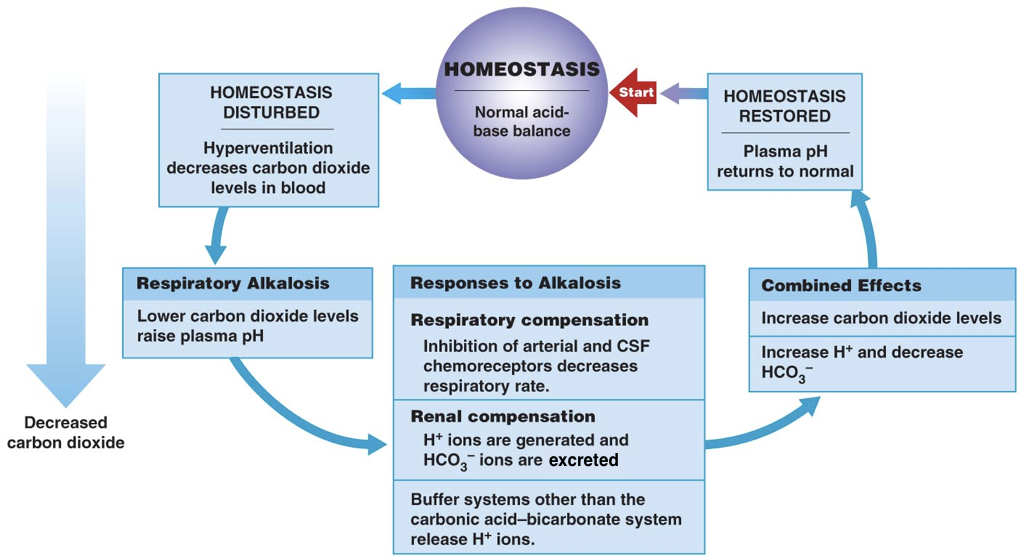
Thus, the pH of blood is an important determinant of the efficiency of oxygen delivery and carbon dioxide removal. When the pH deviates from normal, there are both respiratory and renal compensation mechanisms.
In the respiratory system, the signal to breath is generated by the medulla (the brain stem) in response to rising concentrations of carbon dioxide. In other words, breathing occurs in response to high levels of carbon dioxide rather than low levels of oxygen. Thus, the detection of increased carbon dioxide levels (which lowers the pH) in the blood drives increased respiration, enabling the exhalation of excess carbon dioxide (which restores the pH). In contrast, in response to low levels of carbon dioxide (which raises the pH), there is a decreased respiratory rate, which enables carbon dioxide to accumulate and restore the pH.
In addition to all their other roles, the kidneys can regulate the pH by tempering the blood. In response to acidosis, the kidneys generate and secrete bicarbonate ions (which raise serum pH) and excrete protons (which also raises serum pH). In contrast, in response to alkalosis, the kidneys generate and secrete protons (which lowers the pH) and excrete bicarbonate ions (which also lowers the pH). Together, respiratory compensation and renal compensation for acidosis and alkalosis allow normal oxygen delivery and carbon dioxide removal by red blood cells.
Feedback/Errata