
8 Cortisol & Relaxin
Cortisol and Relaxin
Cortisol
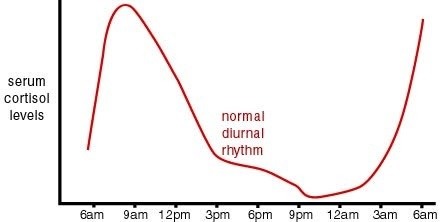 As mentioned previously, glucocorticoids are produced by the zona fasciculata of the adrenal cortex in response to stimulation of MC2R by ACTH (which requires an accompanying MRAP1). ACTH, a section of pro-opio-melanocortin (POMC), is released from the anterior pituitary in response to CRH released into the hypophyseal portal by neurons in the paraventricular nucleus of the hypothalamus. CRH secretion is enhanced beyond basal levels by stress. However, glucocorticoids exhibit a diurnal rhythm in which their levels typically peak just before rising from sleep (which generally occurs just before dawn). This glucocorticoid sinus rhythm is a result of a normal ACTH rhythm. Due to these daily fluctuations in ACTH, it is good that ACTH only has a minimal impact on aldosterone secretion, otherwise aldosterone levels would fluctuate and severely impact fluid and salt balance in the blood stream.
As mentioned previously, glucocorticoids are produced by the zona fasciculata of the adrenal cortex in response to stimulation of MC2R by ACTH (which requires an accompanying MRAP1). ACTH, a section of pro-opio-melanocortin (POMC), is released from the anterior pituitary in response to CRH released into the hypophyseal portal by neurons in the paraventricular nucleus of the hypothalamus. CRH secretion is enhanced beyond basal levels by stress. However, glucocorticoids exhibit a diurnal rhythm in which their levels typically peak just before rising from sleep (which generally occurs just before dawn). This glucocorticoid sinus rhythm is a result of a normal ACTH rhythm. Due to these daily fluctuations in ACTH, it is good that ACTH only has a minimal impact on aldosterone secretion, otherwise aldosterone levels would fluctuate and severely impact fluid and salt balance in the blood stream.
Cortisol is the primary glucocorticoid, and is often referred to as a “stress hormone.” Among their diverse effects, glucocorticoids released into circulation exert negative feedback upon the hypothalamus and anterior pituitary. Due to cortisol’s reactivity, it is usually transported through the blood stream by cortisol-binding globulin (CBG; also called transcortin) or by albumin. This carrier-mediated transport also prevents glomerular filtration, enabling cortisol to remain in circulation. However, bound cortisol requires receptor-mediated transport to enter target cells. Nevertheless, about 4% of circulating cortisol is unbound, and can freely travel into target cells. However, free cortisol is subject to filtration by the kidneys. Cortisone, on the other hand, can freely travel into target cells in the hippocampus, cerebral cortex, liver, and adipose tissue, wherein there is a high expression of 11β1-HSD (also called cortisone reductase) colocalized with cortisol receptors. 11β1-HSD converts the inactive cortisone to active cortisol. The liver has the highest expression of 11β1-HSD. When active cortisol is present in the cytoplasm of cells, it is able to bind to cytoplasmic receptors. This receptor complex can then travel to the nucleus and exert large-scale changes in gene transcription and protein synthesis. In other words, target cells can regulate the effects of glucocorticoids by regulating the availability of active cortisol.
The importance of this auto-regulatory capability is evidenced in cells expressing mineral corticoid receptors (aldosterone receptors). In fact, cortisol has a similar affinity for mineral corticoid receptors as aldosterone, and elicits the same effects. However, cells expressing mineral corticoid receptors (e.g., kidneys, liver, lungs, salivary glands, certain neurons, etc.), also express 11β2-HSD, which is co-localized with mineral corticoid receptors and converts active cortisol into cortisone; cortisone cannot activate mineral corticoid receptors. This prevents the activation of mineral corticoid receptors by cortisol.
In neurons, cortisol tends to increase calcium permeability through voltage-gated calcium channels either directly or indirectly. In the short term, this enhances the ability to cope with stressful stimuli by enhancing excitability, but chronically high cortisol levels can lead to calcium excitotoxicity.
Cortisol regulates carbohydrate metabolism, protein catabolism, and inflammation. These effects tend to suppress the activity of the immune, digestive, and reproductive systems. Cortisol participates in glucose regulation by driving gluconeogenesis.
The initial response to stress is an increased secretion of catecholamines (epinephrine, norepinephrine, and dopamine) by the adrenal medulla in response to sympathetic innervation. Sustained stress increases the production of cortisol through the HPA axis. Together, cortisol and epinephrine drive glycogenolysis through activation of glycogen phosphorylase. This means that if cortisol levels are initially high, a rapid increase in epinephrine can have more profound effects on glycogenolysis. However, without cortisol’s permissive action, catecholamines cannot drive glycogenolysis.
Normally, ACTH and cortisol exhibit a diurnal sinus rhythm in response to the cyclic release of CRH. This causes ACTH and cortisol levels to peak around sunrise, fall throughout the day to a minimum after sunset, and then rise throughout the night. Since night time is generally a period of fasting, rising cortisol at night has an important role in maintaining glucose homeostasis. As insulin levels fall and glucagon levels rise during nightly periods of fasting, there is an increased release of energy stores by the liver (the primary source of fuel during nightly fasting). Rising levels of glucagon initially drive glycogenolysis until glycogen stores are depleted, at which point glucagon drives gluconeogenesis (the conversion of substrates such as β-hydroxybutyrate and acetoacetate into glucose), and ketogenesis (the breakdown of fatty acids into ketone bodies). As glucose levels fall and glucagon levels continue to rise, there is an increased activation of the HPA axis, leading to a rise in cortisol levels. In addition to its promotion of glucose release and impairment of insulin signaling, rising cortisol also promotes the production of ketone bodies as an alternative fuel source. Ketone bodies are small and water-soluble. They can travel freely without carrier proteins, can easily penetrate tissues, and are capable of crossing the blood-brain-barrier. However, due to their small size, they are filtered by glomeruli in the kidney, and due to their strong acidity, they create a large osmotic load, which increases urine production. This increased production of acidic urine in response to rising cortisol contributes to waking in the morning. In addition to increasing glucose availability, glucocorticoids also increase blood pressure, which helps compensate for the loss of blood volume during the night as blood is filtered into the bladder by the kidneys. Since glucocorticoids impair insulin signaling, high stress can also impair insulin signaling and precipitate type II diabetes mellitus.
Glucocorticoids inhibit immune responses at multiple levels. Glucocorticoids suppress immune responses by interfering with autocrine and paracrine signaling in immune cells, including interference with interleukins (IL), interferon (IFN)-γ, IFN-α, and tumor-necrosis factor (TNF)-α. Though immune cells respond to both systemic and local signals, paracrine signaling between immune cells is required for the initiation of large-scale immune responses.
Glucocorticoids significantly diminish the number of circulating lymphocytes (mainly T-cells), basophils, eosinophils, and macrophages (and their monocyte precursors). Lymphocyte counts are decreased through the redistribution of circulating lymphocytes to organs such as the spleen, lymph nodes, and bone marrow. Glucocorticoid-mediated decreases in macrophage and eosinophils are also attributed to redistribution. In addition to reducing the numbers of circulating macrophages, glucocorticoids directly inhibit the activity of macrophages by preventing their ability to respond to chemotactic signals. This is achieved by preventing macrophages from secreting collagenase and elastase (thereby inhibiting collagen and elastin degradation, respectively) and by blocking plasminogen activation (which reduces destruction of blood clots). Glucocorticoids also indirectly impair immune responses by preventing antigen presentation to lymphocytes. Glucocorticoids also completely block IL-2 signaling between T-cells, which prevents the ability of T-cells to stimulate proliferation of other T-cells. So, in addition to the redistribution of T-cells, there is reduced proliferation of new T-cells. In contrast to the glucocorticoid-mediated decreases in some circulating immune cells, glucocorticoids cause an increase in the number of circulating neutrophils. However, glucocorticoids impair neutrophil adhesion to blood vessel walls, which impairs their ability to congregate at inflammatory sites. Furthermore, glucocorticoids also down-regulate the production of prostaglandins by blocking IL-1 production, which would otherwise signal prostaglandin synthesis.
By interfering with immune cell signaling, reducing immune cell counts, and impairing the production of pro-inflammatory signals, glucocorticoids can exert significant suppression of the immune response, endowing glucocorticoids with anti-inflammatory properties. As such, glucocorticoid analogs serve as candidate treatments for asthma (in the case of severe exacerbations), chronic obstructive pulmonary disease (COPD), allergic rhinitis, dermatitis, hives, angioedema, anaphylaxis, and common food and drug allergies.
Cortisol also exerts a significant influence on digestion. While cortisol alters motility through interactions with enteric smooth muscles, cortisol’s impact on microbiota regulation is of particular importance. The microbiota in the digestive tract is both necessary and potentially dangerous. While it is necessary to have a healthy population of bacteria residing in particular regions of the gut, bacteria in the wrong places of the gut can be problematic. For example, Helicobacter pylori (H. pylori) in the stomach are implicated in the development of ulcers. More dangerous is the potential for bacteria in the gut to enter the body (remember that the alimentary canal is external to the body). Normally, the entrance of bacteria is prevented by a population of immune cells serving as sentries along the periphery of the digestive tract. Cortisol both alters the ability of the immune cells to protect the body from gut microbiota, and directly interacts with the microbiota. Cortisol can act as both a signal molecule and energy source for bacteria, and generally alters protein catabolism and release of amino acids and amino acid-derived signal molecules (e.g., serotonin). The uptake of these signal molecules into the bloodstream enables endocrine communication from microbiome to the brain. However, afferent fibers of the vagus nerve receiving enteric signals also provide a direct pathway through which microbiota can influence the brain. Together, these signals produced by bacteria in response to cortisol can have profound impacts on neural processes, such as those contributing to mood.
Cortisol also alters the effectiveness of the HPT axis. Low levels of glucocorticoids increase the effectiveness of the HPT axis, whereas high levels (e.g., during periods of high stress) impair the synthesis of thyroid hormones. Glucocorticoids can also inhibit the effects of thyroxin without affecting circulating thyroxin levels by inhibiting deiodinases, which prevents the conversion of T4 to T3 within target cells.
Due to the variety of effects, it is important that cortisol is maintained within a discrete range, despite its diurnal sinus rhythm. Deviations from this norm in either direction yield significant problems. Addison’s disease, also called primary adrenal insufficiency, is caused by a failure of the adrenal glands to produce sufficient amounts of cortisol. This lack of cortisol results is weakness, weight loss, blood sugar imbalances, fatigue, sweating, nausea, and darkening skin in certain regions. Counter intuitively, Addison’s is characterized by high ACTH due to the lack of negative feedback to the hypothalamus and pituitary by cortisol. This is similar to cases of hypothyroidism stemming from an inability to adequately produce thyroid hormones in response to TSH. Without adequate production of thyroid hormones, there is a lack of negative feedback on TRH and TSH recreation, yielding elevated levels of circulating TSH.
The converse of Addison’s is Cushing’s syndrome, in which abnormally high levels of cortisol are sustained throughout the day (i.e., there is only a minimal diurnal sinus rhythm). While Cushing’s syndrome can arise from Cushing’s disease, it can also result from other causes. In other words, Cushing’s disease leads to Cushing’s syndrome, but not all Cushing’s syndrome is caused by Cushing’s disease. Cushing’s disease is most often caused by pituitary gland dysfunction (e.g., pituitary adenoma) that results in elevated levels of both ACTH and cortisol. It can even result from small-cell pulmonary or hepatic carcinomas that produce ACTH. Interestingly, this means that high ACTH is present in both Addison’s and Cushing’s disease, despite having low and high cortisol levels, respectively. Cushing’s syndrome is characterized by high blood pressure, weakness, rapid weight gain, weak bones, fat accumulation in discrete areas (e.g., the gut and between the shoulders but not extremities), insomnia, and irritability. High levels of psychological stress, which lead to elevated cortisol, can lead to the development of Cushing’s syndrome.
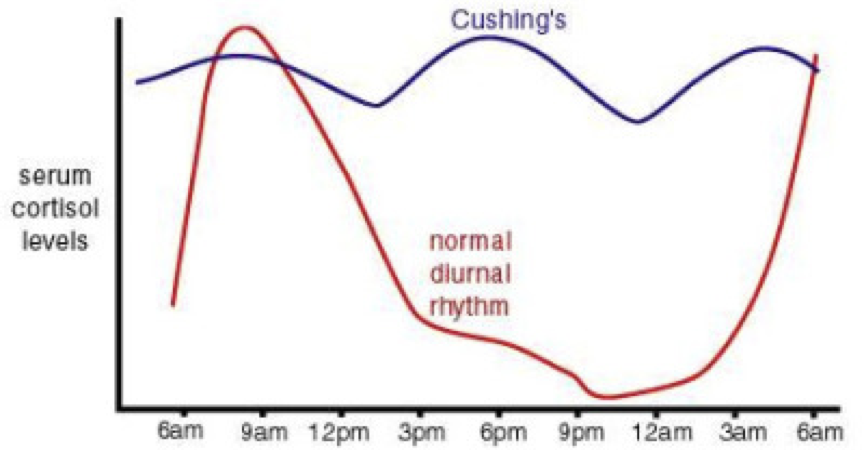
The symptoms of Cushing’s syndrome may also present during corticosteroid treatment (e.g., prednisone used for its immunosuppressant effects). However, acute removal of corticosteroid treatment can precipitate Addison’s due to overstimulation of the adrenal glands by elevated ACTH. Likewise, any other condition, such as perceived stress, can precipitate the symptoms of Cushing’s by, among other things, increasing cortisol secretion. For example, a deficiency in 17, 20-lyase yields an underproduction of adrenal sex steroids, accompanied by an overproduction of cortisol. The converse of this is a 21-hydroxylase deficiency (e.g., in congenital adrenal hyperplasia), which will lead to an underproduction of glucocorticoids and an overproduction of sex steroids (possibly contributing to certain forms of precocious puberty). This is important because chronic stress can actually be fatal, evidenced by animal models. When subordinate male baboons are caged with a dominant male, their chronic stress will lead to the development of gastric ulcers, colitis, enlarged adrenal glands, extensive degeneration of the hippocampus, and likely death if the subordinate males are exposed to the dominant males for a long enough period of time. These effects of chronic stress resemble an accelerated rate of aging processes.
Relaxin
Relaxin, of which there are three types (relaxin 1, 2, and 3), is a hormone structurally related to insulin. Like insulin, relaxin is composed of two peptides of 24 and 29 amino acids, though the sequences are distinct. Nevertheless, it is part of the insulin-like peptide superfamily. Relaxin affects bones, muscles, and connective tissues by inhibiting collagen synthesis, enhancing collagen degradation by proteolytic enzymes, enhancing angiogenesis, and promoting renal vasodilation. Relaxin binding to one of the four types of relaxin receptors (RXFP1, 2, 3, and 4) can also have anti-inflammatory effects, and some forms promote muscle repair while preventing hypertrophic growth. However, relaxin can also weaken ligaments. In fact, increased levels of relaxin increase susceptibility to tearing the anterior cruciate ligaments (ACL).
Estrogen and relaxin have opposite effects on bone production. Estrogen inhibits the NFκB pathway (activated by glucocorticoids) that would otherwise promote osteoclastogenesis. Estrogen promotes OPG production, which preserves bones by inhibiting osteoclasts. In contrast, relaxin appears to inhibit OPG production, which ultimately leads to a net removal of bone material. Despite their antagonistic relationship, both estrogen and relaxin, along with cortisol, are a useful treatment for the symptoms of rheumatoid arthritis when they are injected together into arthritic joints. Cortisol serves as an immunosuppressant, and estrogen promotes collagen production. Despite antagonizing the effects of estrogen to a degree, relaxin increases joint motility by removing certain proteins, such as elastin, which loosens the connections between bones. Treatment with all three hormones is more effective at increasing joint motility than just cortisol and estrogen.
In non-pregnant females, relaxin is produced to a small degree by the breasts, but is predominantly synthesized in the corpus luteum in response to estrogen and progesterone. This causes circulating levels of relaxin to peak during each luteal phase. As a result, the risk for ligament damage changes over the course of a menstrual cycle in proportion to relaxin levels. Taking exogenous estrogen and progesterone (e.g., as a contraceptive) can increase relaxin levels and consequent associated risks. In pregnant females, relaxin is also produced in by the placenta, and peaks both during the first trimester, as well as during parturition. During pregnancy, rising relaxin levels relax the cervix and uterine muscles, softens the pubic bone and enables modification of the pelvic girdle, all of which contribute to a successful birthing process. In males, relaxin is produced in the prostate and to a limited degree in the testis. Relaxin in semen appears to promote sperm motility.
Feedback/Errata