
6 Reproductive Endocrinology Continued I
Endocrine Regulation of Reproductive Systems (Continued)
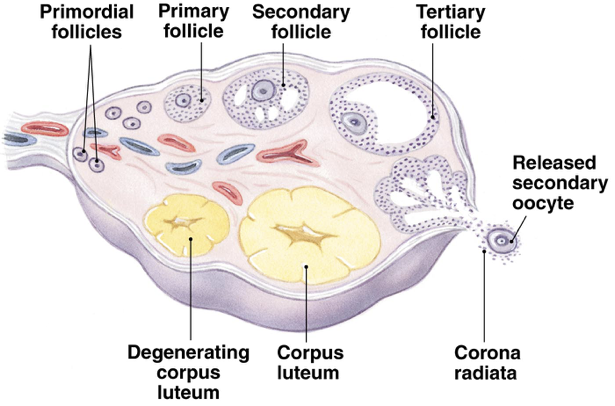
Folliculogenesis and Selection of the Dominant Follicle
Folliculogenesis begins with recruitment of a primordial follicle and ends with ovulation. Throughout the course of folliculogenesis, follicles progress through multiple stages of development necessary to enable successful ovulation of a dominant follicle.
All primordial follicles in the ovaries are formed between about 11 and 20 weeks of gestation. These primordial follicles form the ovary reserve (i.e., the total number of primordial follicles at any given time). At about 20 weeks of gestation, the ovary reserve is at a peak concentration of about 5-million primordial follicles. However, there is a rapid decline in follicle count during the remainder of pregnancy, yielding only an average of about 295,000 primordial follicles at birth (this is extremely variable between individuals though; some females are born with more than 1-million follicles). Over the first four decades of life, the ovarian reserve is gradually depleted. However, after roughly 37.5 years, or when ovarian reserves drop below about 25,000 primordial follicles, the rate at which follicles are depleted doubles. Despite the large number of follicles present at birth, only about 400 primordial follicles will reach the preovulatory stage of folliculogenesis.
Following the initial recruitment of a primordial follicle, folliculogenesis can be broadly divided into two stages: the preantral stage and the antral stage. Development during the preantral stage is independent of gonadotropins, but also extremely slow. The antral stage is dependent on gonadotropin signaling and progresses at a faster rate.
As long as the ovaries contain follicles, local signaling (either autocrine or paracrine) will drive the initial recruitment of primordial follicles. A primordial follicle has only a single layer of flat granulosa cells surrounding the oocyte. Since primordial follicles have no direct blood supply, endocrine signals have minimal impact on primordial follicle development. During transition to a primary follicle, a glycoprotein polymer forms around the oocyte, separating it from the surrounding granulosa cells. This protective glycoprotein layer is called the zona pellucida. The flat granulosa cells of the primordial follicle also divide and enlarge to become cuboidal (cube-shaped) granulosa cells. The cuboidal granulosa cells immediately surrounding the zona pellucida will later comprise the corona radiata, which remains attached to the oocyte throughout ovulation and fertilization.
As granulosa cells develop their cuboid structure, they also begin to express FSH receptors. However, their development persists independent of gonadotropins. Autocrine or paracrine activin signaling by granulosa cells may be responsible for the induction of FSH receptors on primary follicle granulosa cells. Despite expressing FSH receptors, primary follicle granulosa cells are never exposed to concentrations of FSH sufficient for receptor activation. This is primarily because primary follicles lack vasculature and therefore receive no direct exposure to blood-borne signals.
 This transition of a primordial follicle into a primary follicle can take longer than 150 days. The slow rate of development in the preantral stage is largely attributable to the slow doubling rate of granulosa cells, each of which can take longer than 10 days to divide.
This transition of a primordial follicle into a primary follicle can take longer than 150 days. The slow rate of development in the preantral stage is largely attributable to the slow doubling rate of granulosa cells, each of which can take longer than 10 days to divide.
The primary follicle oocyte secretes paracrine signals that drive the transition of the primary follicle into the secondary follicle. Growth differentiation factor 9 (GDF-9) secreted by the oocyte is required for the progression from the primary stage to the secondary stage. Without GDF-9, folliculogenesis is arrested at the primary stage. GDF-9 is a member of the TGF-β superfamily (which also includes inhibin, activin, and AMH). GDF-9 was previously considered to be expressed exclusively in oocytes, but it turns out that GDF-9 is also expressed in anterior pituitary gonadotropes. In anterior pituitary gonadotropes, GDF-9 released as an autocrine or paracrine signal, has effects similar to those of activin. GDF-9 and GnRH operate synergistically to induce the expression of FSH.
Granulosa cells continue to divide and form a second granulosa cell layer; all granulosa cells express FSH receptors. Stroma-like theca cells are recruited around the basal lamina. A capillary network develops to connect the theca cells to the granulosa cell layers. The granulosa cells undergo further division to form more layers. At the end of this secondary stage of development the secondary follicle contains a fully-grown oocyte surrounded by the zona pellucida, up to nine layers of granulosa cells, the basal lamina, and the theca cell layer.
Tertiary follicle development begins when the secondary follicle initiates the process of cavitation, in which fluids accumulate between granulosa cells. Fluids between granulosa cells accumulate gradually and create a cavity within the follicle. The process of cavitation is not reliant on gonadotropins. Instead, it appears that autocrine or paracrine signaling is responsible for the development of the cavity. Two growth factors are particularly implicated in the autocrine or paracrine regulation of cavitation: activin and KIT ligand. In fact, cavitation cannot occur in the absence of KIT ligand signaling.
At the same time cavitation occurs, the stroma-like theca cells differentiate into two distinct layers. Firstly, the theca externa is formed by the outermost stroma-like theca cells, which differentiate into smooth muscle and receive autonomic innervation. The theca interna is formed by the inner layer of stroma-like theca cells. Some of these theca interna cells express LH receptors and are exposed to circulating LH through the follicle’s vasculature. LH receptor activation on these theca interna cells drives their differentiation into theca interstitial cells.
Cavitation is likely the induction stimulus responsible for initiating the process of antral follicle development by permitting the entry of FSH into the follicular fluid. This is also the point at which granulosa cells begin responding to FSH. Activation of granulosa cells by FSH drives further expansion of the cavity, forming the fluid-filled antrum. Once the antrum has developed, the follicle is considered an antral follicle. The process of preantral follicular development, beginning with primordial follicle recruitment, takes about 300 days to complete. An additional roughly 50 days are required for an antral follicle to develop into a preovulatory follicle.
By the time a follicle becomes an antral follicle, most of the follicles that began growth at the same time have already undergone atresia (follicle death), characterized by radical apoptosis of all follicle cells and the oocyte. Further development of the antral follicle is gonadotropin-dependent (primarily dependent on FSH, which both enables further development and prevents atresia). However, both LH and FSH are important for the regulation of antral follicle steroidogenesis, which influences the process of folliculogenesis.
Normal folliculogenesis requires intra-ovarian androgens, which are the precursors for the synthesis of estradiol. However, there is a fine balance between androgens and estrogens. Disruption in the balance impairs follicle maturation and increases follicular atresia. Circulating levels of androgens and estrogens derived from other tissues influence the intra-ovarian balance of androgens and estrogens, and therefore have the potential to influence folliculogenesis.
In normal reproductive-aged females, theca cells (of the theca interna layer) of ovarian follicles and the adrenal cortex contribute equally to levels of circulating androgens. Both secrete more androstenedione than testosterone. LH (at theca cells) and ACTH (in the adrenal cortex) are responsible for inducing production of these androgens.
Androstenedione is metabolized in peripheral tissues (primarily the lungs, liver, adipose tissue, and skin). The peripheral metabolism of androstenedione to testosterone is responsible for almost half of testosterone in normal reproductive-aged females. Androstenedione is also aromatized into estrone, largely in adipocytes. Almost all circulating DHT in females is also produced in peripheral tissues, predominantly from circulating androstenedione through a two-step conversion; first to testosterone, and then to DHT. Androgen levels in healthy females undergo an approximately twofold change depending on the diurnal rhythm and menstrual cycle.
LH binding to theca cells in the ovary (and ACTH binding to cells of the adrenal cortex) stimulates the activity of CYP17A1. With its 17α-hydroxylase activity, CYP17A1 converts pregnenolone to 17α-hydroxypregnenolone, and with its 17, 20-lyase activity, CYP17A1 converts 17α-hydroxypregnenolone to dehydroepiandrosterone (DHEA). LH in the ovary and ACTH in the adrenal cortex also stimulate a parallel pathway, in which pregnenolone is converted to progesterone by 3β-HSD, and then, through its 17α-hydroxylase activity, CYP17A1 converts progesterone to 17α-hydroxyprogesterone (17-OHP). DHEA can be converted to androstenedione by 3β-HSD, and 17-OHP can be converted to androstenedione by CYP17A1 due to its 17, 20-lyase activity. However, some 17-OHP in the adrenal cortex is converted to 11-deoxycortisol by 21-hydroxylase, and then to cortisol by 11β-hydroxylase. Androstenedione requires the activity of 17β-HSD for conversion to testosterone, which can then be converted to dihydrotestosterone or estradiol.
This LH-stimulated androgen production occurring in theca cells (primarily yielding androstenedione) is required for the production of estrogens by granulosa cells. This is characterized as the two-gonadotropin, two-cell theory, in which LH is responsible for inducing theca cells production of androstenedione from cholesterol, and FSH is responsible for inducing aromatization of androstenedione into estrone, and then conversion to estradiol by 17β-HSD. Androgen synthesis in theca cells is directly correlated with aromatase activity in granulosa cells. In healthy follicles greater than 8mm in diameter, androstenedione from theca cells is efficiently converted to estradiol by granulosa cells. The steroidogenic activities of theca cells and granulosa cells are coordinated by autocrine, paracrine, and endocrine signals.
In addition to FSH and LH as endocrine signals regulating steroidogenesis, paracrine activin, inhibin, and anti-müllerian hormone (AMH) are also very important. The combination of FSH, LH, activin, inhibin, AMH, and their effects on steroidogenesis, collectively interact to determine which follicle will be selected to progress to the preovulatory stage (i.e., they determine which follicle will be selected as the dominant follicle).
Selection of the dominant follicle begins in the late-luteal phase of each menstrual cycle. If the oocyte released at the mid-cycle LH surge was not fertilized and did not implant, then the corpus luteum formed from that oocyte’s ruptured follicle will begin to degrade at the end of the luteal phase. The resultant decrease in circulating estrogen, progesterone, and inhibin will release inhibition on FSH secretion, leading to an increased secretion of FSH in the late-luteal phase. FSH levels peak in the early-mid-follicular phase (this peak is not as substantial as the mid-cycle FSH peak that accompanies the LH surge). The decrease in estradiol in the late-luteal phase following degradation of the corpus luteum seems to be the most important factor enabling the increase in FSH.
Circulating levels of AMH and inhibin indicate the total number of remaining follicles, and can be used to predict the onset of menopause (i.e., the more follicles, the more AMH and inhibin; the fewer follicles, the less AMH and inhibin). Low levels of AMH are also associated with an increased likelihood of producing dizygotic twins.
To efficiently compete for the FSH, follicles increase their exposure to FSH, and their responsiveness to FSH. Follicles concentrate FSH in their follicular fluid. Follicles selected as dominant follicles are found to have relatively high concentrations of FSH in their follicular fluid compared to very low or even undetectable concentrations of FSH in the follicular fluid of nondominant follicles destined for atresia. FSH stimulates aromatase expression in granulosa cells (thereby stimulating estradiol production), but also stimulates the release of inhibin and AMH from granulosa cells. Inhibin enhances LH-stimulated production of androgens (through antagonism of activin’s inhibition of androgen synthesis), and AMH inhibits the production of estradiol by granulosa cells (i.e., AMH antagonizes the effects of FSH). However, the temporal pattern of expression in response to FSH differs between these two paracrine hormones.
During the late-luteal phase AMH levels increase due to increased FSH, but during the early-follicular phase, AMH expression decreases as the follicle grows, despite increases in FSH signaling; once a follicle reaches about 9mm in diameter, AMH production is significantly decreased (this may be due to increased estrogen-mediated inhibition of AMH expression). AMH also seems to have a significant role in the inhibition of primordial follicle recruitment and earlier stage development, in which inter-follicular AMH inhibits development, preventing recruitment and development of an excessive number of follicles, and thereby limiting the rate at which the finite ovarian reserve is depleted. Nevertheless, as intrafollicular AMH decreases during follicular growth, there is a release of inhibition on FSH-stimulated estradiol synthesis, permitting a rise in estradiol.
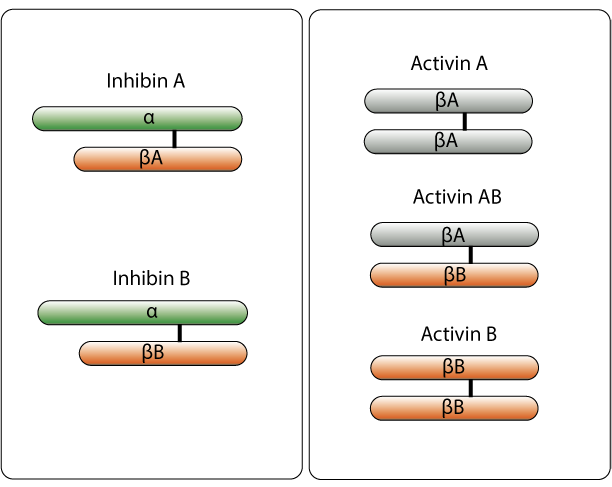 Inhibin exerts its effects not by binding to ‘inhibin receptors,’ but rather by binding antagonistically to receptors for activin (activin receptors; ACVRs). Activin and inhibin both belong to the same cytokine family, the transforming growth factor β (TGF-β) protein superfamily, which also includes hormones such as anti-müllerian hormone (AMH), bone morphogenetic protein, and growth differentiation factor, all of which (including activin and inhibin) are peptide hormones with related structures. To activate intracellular signaling pathways, TGF-β superfamily ligands bind to type II receptors, which recruit and phosphorylate type I receptors. The type I receptors then phosphorylates receptor-regulated SMADs (R-SMADs), which bind to co-SMADs (SMAD4 is the only co-SMAD used in TGF-β superfamily signaling). The R-SMAD/SMAD4 then complex serves as a transcription factor. Activin and inhibin are dimeric structures composed of two monomers linked by a single disulfide bond. There are two forms of inhibin, both of which are heterodimers. Inhibin A is formed by an α monomer and a βA monomer, and inhibin B is formed by an α monomer and a βB monomer. There are three known forms of activin, which are composed of different combinations of the same β monomers that form inhibin. Activin A is a homodimer formed by two βA monomers, activin B is a homodimer formed by two βB monomers, and activin AB is a dimer formed by one βA monomer and one βB monomer.
Inhibin exerts its effects not by binding to ‘inhibin receptors,’ but rather by binding antagonistically to receptors for activin (activin receptors; ACVRs). Activin and inhibin both belong to the same cytokine family, the transforming growth factor β (TGF-β) protein superfamily, which also includes hormones such as anti-müllerian hormone (AMH), bone morphogenetic protein, and growth differentiation factor, all of which (including activin and inhibin) are peptide hormones with related structures. To activate intracellular signaling pathways, TGF-β superfamily ligands bind to type II receptors, which recruit and phosphorylate type I receptors. The type I receptors then phosphorylates receptor-regulated SMADs (R-SMADs), which bind to co-SMADs (SMAD4 is the only co-SMAD used in TGF-β superfamily signaling). The R-SMAD/SMAD4 then complex serves as a transcription factor. Activin and inhibin are dimeric structures composed of two monomers linked by a single disulfide bond. There are two forms of inhibin, both of which are heterodimers. Inhibin A is formed by an α monomer and a βA monomer, and inhibin B is formed by an α monomer and a βB monomer. There are three known forms of activin, which are composed of different combinations of the same β monomers that form inhibin. Activin A is a homodimer formed by two βA monomers, activin B is a homodimer formed by two βB monomers, and activin AB is a dimer formed by one βA monomer and one βB monomer.
Starting in the late-luteal phase (in response to the late-luteal phase rise in FSH), inhibin-B levels increase, peaking in the mid-follicular phase one- to two-days after the inter-cycle FSH peak. Inhibin-B from granulosa cells down-regulates FSH from the anterior pituitary (a classic case of negative feedback), and enhances androgen synthesis in theca cells. In addition to providing precursors for estrogens, the increased production of androgens induces increased FSH receptors (and LH receptors) on granulosa cells. Thus, the granulosa cells receive a stronger signal to produce estrogens (due to increased FSH receptors), and have the necessary precursors available. Without AMH to restrain production, estradiol production is significantly enhanced. At this point, FSH levels have already begun to decrease due to negative feedback from inhibin-B. However, the rise in estradiol provides further negative feedback. The follicle that (due to high production of inhibin-B) expresses the most FSH (and LH) receptors will be able to withstand the decreased exposure to FSH, and will therefore emerge as the dominant follicle. The competing follicles (without sufficient FSH receptors) will become atretic due to decreased stimulation by FSH.
Thus, it appears that follicles compete for dominance by racing to increase FSH concentrations in their follicular fluid, to increase their responsiveness to FSH, and to impair the development of competing follicles by reducing FSH secretion by the anterior pituitary. The dominant follicle selected through 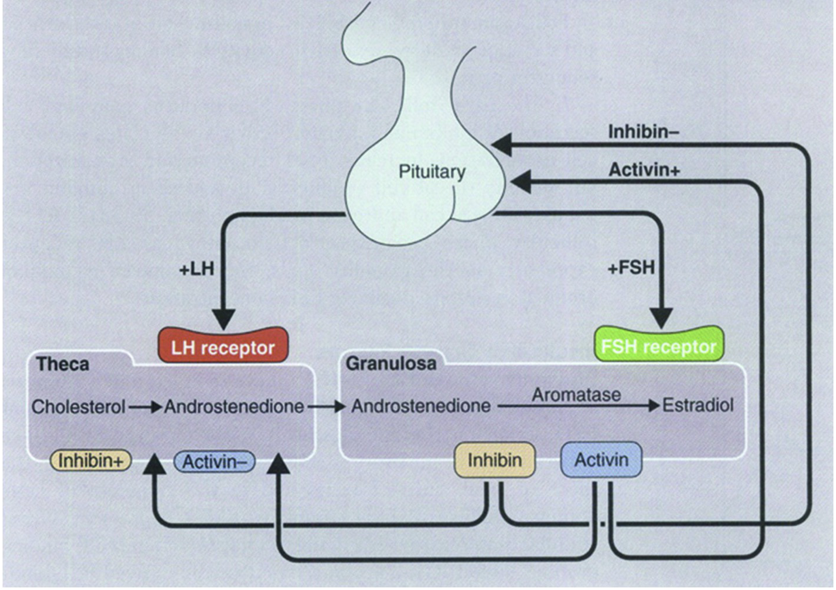 this process would exhibit the most favorable pattern of receptor expression and the most efficient vascularization. This ensures that the selected follicle will be capable of growing rapidly into a preovulatory follicle. This ensures that the dominant follicle capable of developing into the preovulatory follicle.
this process would exhibit the most favorable pattern of receptor expression and the most efficient vascularization. This ensures that the selected follicle will be capable of growing rapidly into a preovulatory follicle. This ensures that the dominant follicle capable of developing into the preovulatory follicle.
Selection of the dominant follicle usually occurs on day 7 of the menstrual cycle (i.e., in the mid-follicular phase). The dominant follicle will continue to rapidly develop, growing up to 20mm in diameter. Due to the induction of LH receptors on granulosa cells, the dominant follicle is capable of responding to the mid-cycle LH surge, and capable of releasing its oocyte during ovulation.
Inhibin’s antagonism of activin is primarily achieved through competitive binding to the activin type II receptors with the shared β-subunit. Inhibin binding to activin type II receptors prevents recruitment of activin type I receptors. Instead, activin type II receptors, when bound by inhibin, recruit betaglycan, which prevents signaling. Activin has multiple roles in both males and females. Activin is primarily used as an autocrine or paracrine signal. Activin and inhibin are produced in the gonads, anterior pituitary gland, placenta, and other organs. As long as stress levels are low and sex steroid levels are high, activin is produced in a high quantity to maintain health. Activin (released as an autocrine signal by gonadotropes, or as a paracrine signal by other anterior pituitary cells) enhances FSH synthesis: when activin binds to receptors on the anterior pituitary, it enhances the effects of GnRH such that the same amount of GnRH will yield a greater change in FSH production; activin and GnRH operate synergistically in stimulating FSH production. In the ovarian follicle, activin enhances FSH binding, androgen synthesis, and consequent aromatization of androgens to produce estrogens. In males, activin enhances spermatogenesis. Though it is also involved in cell proliferation and differentiation, activin is also involved in wound healing. When cells are damaged (particularly skin and muscle cells), they express certain activin receptors on their surface. They can then release activin as an autocrine signal to undergo apoptosis. Importantly, other cells will not be affected because they will lack the appropriate types of activin receptors. Activin is strongly expressed in wounded skin, and in transgenic mice, its overexpression has been shown to improve wound healing and to enhance scar formation. Activin levels are elevated in the liver following removal of a lobe for transplantation, participating in the subsequent growth and repair. Though these are not all of the roles of activin, one particularly important role is activin’s promotion of insulin secretion by β-cells (comparable to the effects of GLP-1); activin has an incretin effect. Furthermore, activin may even enhance β-cell proliferation. Due to the importance of activin’s diverse roles, mutations that disrupt this system can have dramatic effects. A mutation in the activin type I receptor (ACVR1) results in a condition called fibrodysplasia ossificans progressiva (FOP), a fatal disease that causes muscle and soft tissue to gradually ossify (turn to bone). The presence of the disease is easily identifiable in infants due to abnormal toes. While the ACVR1 gene normally signals healing, mutations can signal ossification. Fortunately, a drug has been developed that aids in the treatment of FOP. In contrast to activin, inhibin is commonly used as an endocrine hormone, and inhibits FSH production. Inhibin in males and females can be secreted by the anterior pituitary. In males, inhibin is also secreted from Sertoli cells, and in females, inhibin is also secreted by granulosa cells of ovarian follicles and corpus lutea, as well as by the placenta during pregnancy. Inhibin binds to activin receptors on the anterior pituitary at the activin binding site, preventing the action of activin. This prevents activin-mediated increases in FSH synthesis, yielding a functional reduction in FSH levels.
Pregnancy
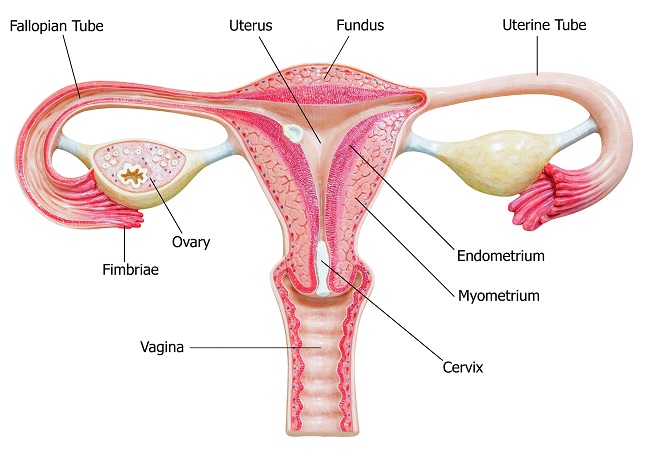
After an oocyte is released during ovulation, it must enter a fallopian tube via fimbria. In the fallopian tubes, mobile cilia capable of transporting the oocytes through the fallopian tubes to the uterus carry the oocyte. However, this transportation by fallopian tube cilia takes about seven days. This is problematic because there is a limited, roughly 24-hour post-ovulatory period during which the oocyte is capable of being fertilized. Due to the slow rate of oocyte transport through fallopian tubes and the limited time-frame during which the oocyte can be fertilized, the majority of fertilization events occur within the fallopian tubes.
Fertilization
As sperm ascend through the female genital tract, they undergo several physiological changes that enable their penetration of the oocyte. The first change is capacitation, which increases sperm mobility and prepares sperm for the enzymatic reaction necessary to penetrate the oocyte.
Fertilization promoting peptide (FPP) is produced by the prostate gland and is excreted into semen. In the semen, the concentrated FPP inhibits capacitation. However, the concentration of FPP is diluted by vaginal excretions, and the increased pH of the female genital tract following ovulation further reduces the activity of FPP. Removing the inhibitory effects of FPP permits capacitation. The endometrium also secretes sterol-binding albumin, lipoproteins, and proteolytic enzymes.
Spermatozoa are surrounded by sterols (e.g., cholesterol), as well as epididymal and seminal glycoproteins. The secretions by the endometrium remove the sterols and epididymal and seminal glycoproteins, resulting in an increased membrane fluidity and increased calcium permeability. The resultant calcium influx raises intracellular calcium concentrations. Synergistically with adenosine, increased intracellular calcium increases activity of adenylyl cyclase, yielding increased levels of cAMP. The increased production of cAMP leads to hyperactivity, including discharging whipping movements of the flagellum and larger swinging movements of the acrosomal head. The destabilization of the acrosomal head (due to the removal of sterols and glycoproteins) enables the acrosomal reaction necessary to penetrate the outer layers of the oocyte. As a result, capacitation promotes the likelihood of successful fertilization. The importance of capacitation for achieving successful fertilization has implications for in vitro fertilization (IVF). Since capacitation results from diverse processes occurring in vivo, IVF must find ways to replicate those processes.
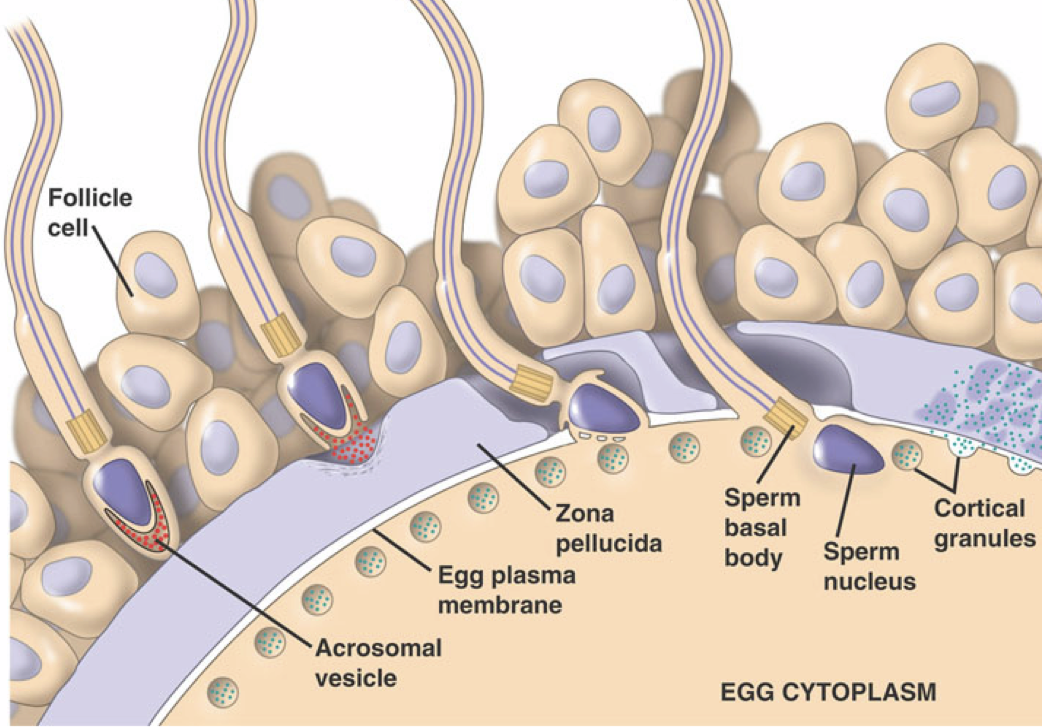
The oocyte membrane is surrounded by the zona pellucida, an extracellular matrix of glycoproteins forming a thick protective layer. The zona pellucida is surrounded by follicle cells forming the corona radiata. When a sperm reaches the oocyte, it must penetrate the layers surrounding the oocyte to achieve successful fertilization.
The sperm passes through the corona radiata, and binds to glycoproteins on the zona pellucida. This binding of glycoproteins induces the release of enzymes from the acrosomal vesicle, which degrade the zona pellucida. This enzymatic penetration of the zona pellucida is the acrosome reaction. The penetration of the zona pellucida induces a sudden depolarization of the oocyte membrane, which initiates an influx of calcium. Among other effects, the calcium influx drives the exocytotic release of cortical granules by the oocyte. The cortical granules contain enzymes that cause glycoproteins of the zona pellucida to link together, preventing penetration of the zona pellucida by other spermatozoa. The acrosomal head also contains the spermatozoa nucleus. Once the nucleus of the sperm and the nucleus of the oocyte fuse, yielding a diploid zygote, fertilization has occurred.
Ectopic pregnancy is a life-threatening condition resulting from the implantation of a fertilized oocyte outside of the uterine endometrium. The vast majority of ectopic pregnancies result from fertilized oocytes implanting in the fallopian tubes (about 95% of all cases), but they can also result from fertilized oocytes implanting in the ovaries, abdominal cavity, or cervix. Ectopic pregnancies are usually recognized within the first 13 weeks of gestation (in the first trimester) due to the presentation of associated symptoms. The Symptomatic presentation often includes severe abdominal pain, vaginal bleeding, nausea, faint feeling, and amenorrhea. However, some cases of ectopic pregnancy are asymptomatic. If ectopic pregnancies are detected early, management may initially involve watchful waiting, since ectopic pregnancies often spontaneously miscarry. Methotrexate (a chemotherapy agent) inhibits rapidly dividing cells and may be taken to stop the growth of the ectopic pregnancy. If a patient with an ectopic pregnancy is hemodynamically unstable, surgery is often performed. If an ectopic pregnancy has ruptured, surgery is always performed because a ruptured ectopic pregnancy is life-threatening. Signs and symptoms of a ruptured ectopic pregnancy include referred pain perceived in the shoulder tip, tachycardia, hypotension, signs of shock, and signs of peritonitis. Surgery for ectopic pregnancies often involves opening the fallopian tube (salpingostomy) to remove the ectopic pregnancy or removal of the entire fallopian tube (salpingectomy).
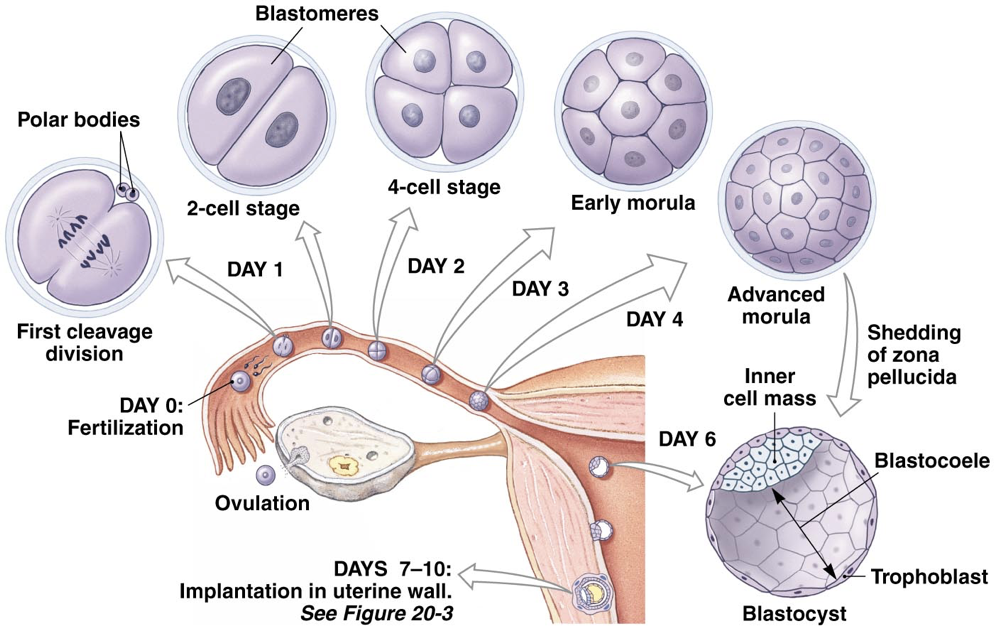
The zygote is transported through the fallopian tube by cilia, usually requiring about seven days to reach the uterus. If fertilization occurs prior to the oocyte entering a fallopian tube or if there is a failure to transport the zygote through the fallopian tube after fertilization, an ectopic pregnancy may result, in which a fertilized egg implants somewhere other than the endometrium of the uterus (most often in a fallopian tube).
As the zygote is moved through the fallopian tube to the uterus, it begins dividing. This process of division is known as cleavage; the number of cells increases, but the size of the cell mass remains the same, enclosed within the zona pellucida. Once there are 16 cells (called blastomeres) clustered together (usually about four days post-ovulation), the cell mass is considered a morula. The blastomeres continue to divide and begin to differentiate. At about six days post-ovulation, the resultant blastocyst contains 32 blastomeres and a fluid-filled cavity called the blasteole. Differentiation of blastomeres from the morula yield two types of blastomeres in the blastocyst. Some blastomeres form the inner cell mass (also called the embryoblast), whereas others, called trophoblasts, form the outer ring of the blastocyst. The inner cell mass is destined to become the three germ layers of the developing embryo (endoderm, mesoderm, and ectoderm) as well as the amniotic sac supporting the developing embryo.
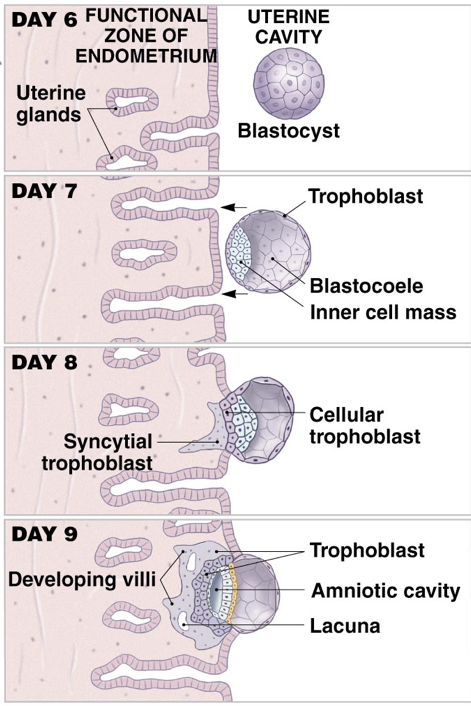
 Prior to implantation, the zona pellucida surrounding the blastocyst breaches, a process called ‘hatching.’ Without the zona pellucida, the size of the blastocyst is no longer constrained, and trophoblasts are exposed to the uterine environment. Trophoblasts secrete autocrine factors as well as enzymes that degrade the endometrial lining. Together, these secretions enable invasion of the endometrium and subsequent implantation, usually occurring about eight- or nine-days post-ovulation.
Prior to implantation, the zona pellucida surrounding the blastocyst breaches, a process called ‘hatching.’ Without the zona pellucida, the size of the blastocyst is no longer constrained, and trophoblasts are exposed to the uterine environment. Trophoblasts secrete autocrine factors as well as enzymes that degrade the endometrial lining. Together, these secretions enable invasion of the endometrium and subsequent implantation, usually occurring about eight- or nine-days post-ovulation.
When the blastocyst implants (ideally in the endometrium of the uterus), the implantation event initiates the release human chorionic gonadotropin (hCG) from trophoblasts, which combine with the maternal endometrium to form the placenta (which continues to secrete hCG). hCG is both a paracrine and endocrine signal. It is a paracrine signal to the endometrium that implantation has occurred, and also has systemic effects. Notably, the secretion of hCG prevents degradation of the corpus lutuem, which maintains high levels of progesterone and prevents the onset of another menstrual cycle. Pregnancy tests work by measuring the presence (or absence) of hCG. Specifically, pregnancy tests use antibodies specific to the hCG β-subunit (hCGβ). Since hCG has the same αGSU as LH and FSH, measuring hCGβ limits the risk of false positive pregnancy resulting from recognition of LH or FSH.
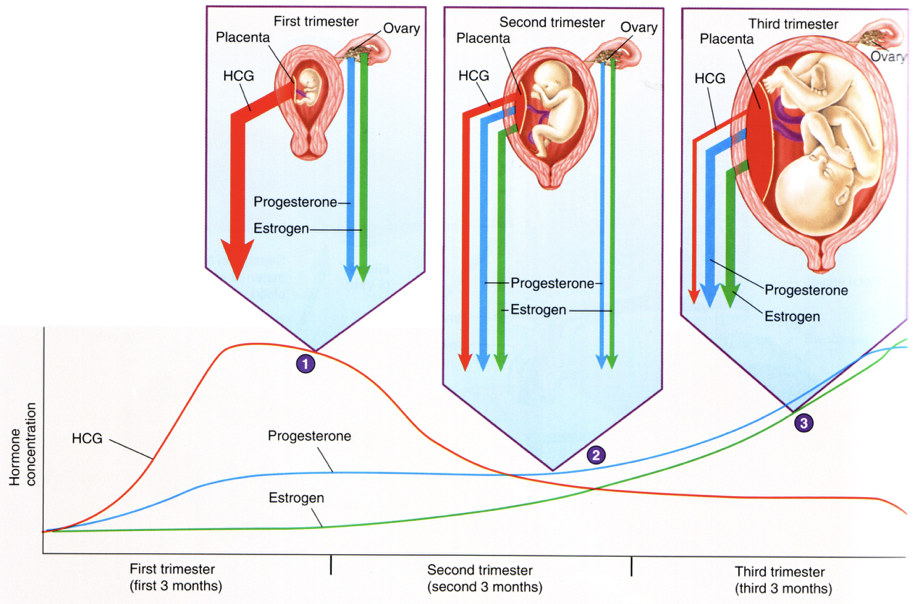
hCG produced by the placenta rises to its peak concentration during the first trimester, but then drops gradually for the remainder of the pregnancy. hCG stimulates the release of estradiol and progesterone throughout pregnancy through stimulation of LHCG receptors, the same receptors activated by LH. Since these receptors are present on the corpus luteum, endocrine hCG signaling elicits a large production of estradiol and progesterone by the corpus luteum during the first trimester, accompanied by low levels of FSH and LH (due to inhibition of GnRH, which was already inhibited by high levels of progesterone and estradiol in the luteal phase). However, as the placenta develops, it begins to respond to paracrine hCG signaling. By the second trimester, the placenta assumes responsibility for the majority of estrogen and progesterone production. This shift in steroid synthesis can be explained by receptor expression. In the first trimester, hCG levels are high, enabling their stimulation of LHCG receptors on the corpus luteum. This coincides with the induction of LHCG receptor expression on the developing placenta. As hCG levels decrease throughout the second and third trimester, there is less stimulation of steroidogenesis in the corpus luteum. However, due to the increasingly efficacious pattern of LHCG receptor expression on the placenta, the decreasing levels of hCG continue to elicit increasing production of placental estrogens and progesterone, yielding a rise in estrogens and progesterone throughout pregnancy.
In addition to the high levels of progesterone characteristic of pregnancy, a female’s progesterone levels can be quite high even when she is not pregnant. If progesterone levels are high while GnRH levels are low, it is most likely during the luteal phase, during which progesterone is produced by the corpus luteum and inhibits GnRH release.
The three peptide hormones, αMSH, βMSH, and γMSH (mentioned previously), also play a role in pregnancy. Each form of MSH is very similar. There is a rise in MSH during pregnancy. As mentioned, POMC is the precursor to all three types of MSH. Since each MSH is cleaved from a different portion of POMC, all three can be produced from a single POMC. However, αMSH requires the same portion of POMC as ACTH (it is actually produced from a portion of the ACTH sequence). MSH has multiple roles, one of which involves the induction of melanin production by skin cells independent of genetic background. This occurs in response to αMSH produced by the anterior pituitary.
Skin pigmentation based on genetic background has different origins. Some people have an MSH receptor deficiency, resulting in red hair, white skin, and freckles (usually most prevalent in northern Europe, e.g., Ireland). In general, though, the changes in skin pigmentation due to MSH are dependent on the environment. During pregnancy, certain regions of the body darken in color due to increased MSH.
However, MSH is also involved in growth regulation. The increase in MSH during pregnancy is theorized to promote the health of the mother to enable a successful pregnancy. MSH also has other roles. αMSH is produced from POMC in the arcuate nucleus of the hypothalamus, where it has roles in appetite suppression and sexual behavior. Due to their common sequence, αMSH and ACTH are both agonists at certain melanocortin receptors (MCRs). Nevertheless, only ACTH stimulates MC2R (Agouti-related protein and agouti-signaling protein are antagonist peptides at MC2R). However, to bind to an MC2R, a melanocortin-2 receptor accessory protein-1 (MRAP1) must also be bound. Without MRAP1, the receptor will never be translocated to the surface of the cell membrane. Rather, MC2R will be degraded in the ER. Thus, by regulating the availability of MRAP1, the activity of ACTH can be regulated. This role of MRAP1 is best exemplified by its effects on daily cortisol cycles (and its consequent effects on daily testosterone cycles). There is a change in circulating testosterone levels over a 24-hour time frame caused by cortisol. There is a cortisol spike prior to waking, followed by a rise in testosterone. MRAP1 expression may be responsible for this daily cortisol cycle, and consequent testosterone cycle.
Another important caveat about MSH synthesis is that the creation of plenty of ACTH means that there is plenty of POMC available for conversion to MSH. For this reason, an excessive amount of ACTH can actually lead to a counterintuitive increase in MSH. This is very different from the steroid synthesis pathways, which compete for the same precursors. For this reason, stress that leads to elevated ACTH can also lead to changes in skin pigmentation. Notably, MCRs can also be blocked by anti-MSH, a competitive inhibitor.
Embryonic Sexual Differentiation
In the first six weeks of embryonic development, male (XY) and female (XX) embryos undergo the same developmental processes. By about six weeks, both male and female embryos possess a cloaca (which becomes the ureters, bladder, urethra, and rectum), Müllerian ducts (the scaffolding for the female reproductive system), Wolffian ducts (the scaffolding for the male reproductive system), and bipotential gonads (capable of differentiating into either testes or 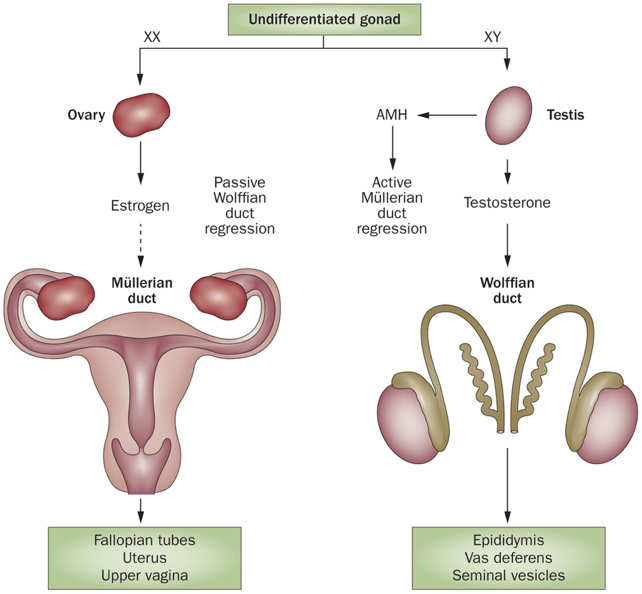 ovaries). However, following about six weeks of embryonic development, sexual differentiation begins.
ovaries). However, following about six weeks of embryonic development, sexual differentiation begins.
In males, the Y chromosome contains the sex-determining region Y (SRY) gene. The SRY gene encodes the SRY protein (also called testes determining factors, TDF), a transcription factor that regulates transcription of other transcription factors. Among other effects, the SRY protein upregulates transcription of the SRY-like box 9 (SOX9) gene, yielding the transcription factor SOX9. SOX9 stimulates the development of the seminiferous tubules from the undifferentiated gonad, leading to formation of the testes, including the development of Leydig cells and Sertoli cells. The Leydig cells begin secreting testosterone, which causes Wolffian ducts to develop into male reproductive structures, including the seminal vesicles, vas deferens, and epididymis.
In chromosomal male embryos, if there is a failure to produce androgens, or a failure in androgen signaling, the Wolffian ducts will degenerate. In other words, the Wolffian ducts will degenerate in the absence of testosterone. At the same time, the Sertoli cells secrete AMH. AMH signals the regression of Müllerian ducts. In fact, AMH will signal the regression of the Müllerian ducts in both male and female embryos (i.e., both express AMH receptors), but only males produce AMH.
In the absence of functional AMH signaling, the Müllerian ducts continue to develop in male and female embryos. In other words, Müllerian duct development is the default pattern of development. The loss of the AMH receptor type 2 (AMHR2) in male fetuses results in an intersex reproductive phenotype with both Wolffian and Müllerian ducts.
In female embryos, which lack the SRY gene, bipotential gonads will develop into ovaries. Without Leydig cells, the lack of testosterone allows the Wolffian ducts to degenerate. Likewise, without Sertoli cells, the lack of AMH allows the Müllerian ducts to develop into the fallopian tubes and uterus. Though AMH is not produced in female embryos, AMH is an important paracrine signal involved in follicle selection later in life.
These changes all occur by about week 10 of embryonic development. In males after about week 10, the testes begin to descend from the abdominal cavity, the cloaca develops into the ureters, bladder, urethra, and rectum. While testosterone is primarily responsible for the development of internal male reproductive structures, DHT seems to be vital for normal development of external male genitalia. Without DHT, external genitalia will resemble female genitalia. By birth, all reproductive structures are present.
The efficacy of normal sex differentiation depends on the presence of appropriate receptors. The hormonal environment influences which receptor types and concentrations are present, most notably within the hypothalamus. As such, a large contributing factor in determining which receptors are present in the hypothalamus is the hormonal environment of the mother during pregnancy. The placental barrier also plays a large role in determining what hormones from the mother are capable of affecting the developing fetus. As the mother’s hormones change throughout pregnancy, the hypothalamus changes as well. Both males and females express both estrogen and androgen receptors, but in different concentrations. Furthermore, males and females produce different sex hormones during development. Interestingly, a substantial amount of male masculinization due to testosterone is induced by estrogen receptor activation following the aromatization of testosterone within the hypothalamus. Interestingly, there are actually sexually dimorphic regions of the hypothalamus, including the third interstitial nucleus of the anterior hypothalamus, and the sexually dimorphic nucleus of the preoptic area.
Parturition
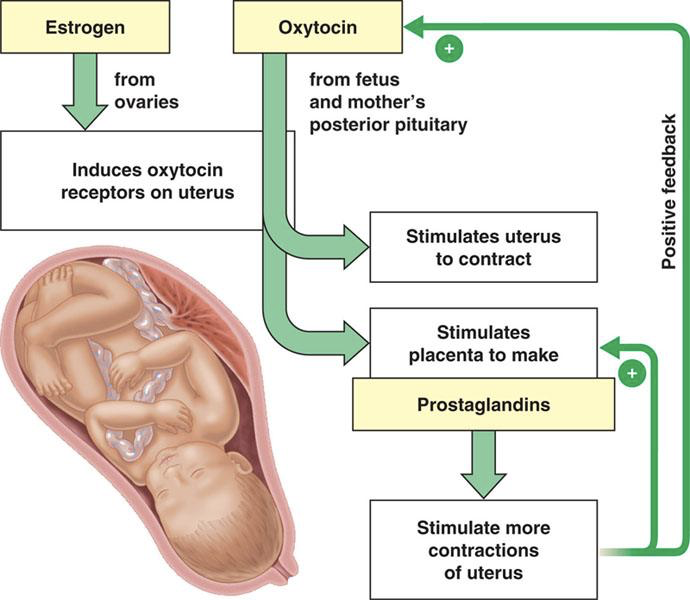 Oxytocin, synthesized in hypothalamic magnocellular neurons and released from the posterior pituitary, plays an endocrine role in childbirth (parturition) and nursing. Oxytocin is also used as a neurotransmitter, and together with the endocrine release of oxytocin, neuronal oxytocin signaling is involved in mother-child bonding. With regards to childbirth, oxytocin is released from the posterior pituitary in response to stimulation of stretch receptors on the cervix and uterus. Oxytocin then stimulates contraction of the myometrium and stimulates the placenta to make prostaglandins. Prostaglandins also stimulate myometrial contractions via paracrine signaling. Uterine contractions stimulate the placenta to produce more prostaglandins, and stimulate stretch receptors that signal a further increase in oxytocin production (both via positive feedback).
Oxytocin, synthesized in hypothalamic magnocellular neurons and released from the posterior pituitary, plays an endocrine role in childbirth (parturition) and nursing. Oxytocin is also used as a neurotransmitter, and together with the endocrine release of oxytocin, neuronal oxytocin signaling is involved in mother-child bonding. With regards to childbirth, oxytocin is released from the posterior pituitary in response to stimulation of stretch receptors on the cervix and uterus. Oxytocin then stimulates contraction of the myometrium and stimulates the placenta to make prostaglandins. Prostaglandins also stimulate myometrial contractions via paracrine signaling. Uterine contractions stimulate the placenta to produce more prostaglandins, and stimulate stretch receptors that signal a further increase in oxytocin production (both via positive feedback).
These are two of the few positive feedback loops. However, there must be high levels of estrogen to induce oxytocin receptors in the uterus, so oxytocin only stimulates uterine contractions at the end of pregnancy when estrogen levels are sufficiently high to induce receptor expression. For this reason, it is also necessary to block oxytocin receptors in the event of a caesarean section birth.
Prostaglandins can be used as both endocrine and paracrine signals, and are distinct from steroid and peptide hormones. Immune cells, such as mast cells, macrophages, and neutrophils, are activated in response to tissue damage. These immune cells release inflammatory mediators, such as prostanoids, bradykinins, and histamine, and recruit other immune cells. Prostanoids and bradykinins bind to prostanoid receptors and B2 receptors, respectively, on nociceptors, which signal pain to the brain. The nociceptors also release inflammatory mediators such as substance P. In addition, damaged tissues themselves release inflammatory mediators. Normally when cells rupture, ATP and potassium inside the cell, and components of the interior lipid bilayer, are exposed to the interstitial space. Prostanoids are one of the most important pro-inflammatory mediators, and serve multiple functions. Of the numerous prostanoids, prostaglandins and thromboxanes are the most important. Prostanoid production begins with the creation of arachidonic acid from diacylglycerol or from phospholipids (e.g., by phospholipase C or phospholipase A2). Arachidonic acid is converted to prostaglandin H2 by cyclooxygenase enzymes (COX1 and COX2). Multiple prostanoids can then be produced from prostaglandin H2. For example, in immune cells, after creating prostaglandin H2 from arachidonic acid, prostaglandin E synthase (PGE) converts prostaglandin H2 into prostaglandin E2. In platelets, COX1 is constitutively expressed, enabling production of prostaglandin H2 from arachidonic acid. Prostaglandin H2 is then converted to thromboxane A2 by thromboxane synthase. Thromboxane A2 aids in platelet aggregation to stop bleeding. COX1 is constitutively expressed, and is involved in homeostatic processes such as clotting, gastric protection, and renal function. In contrast, COX2 is inducible by physiological trauma, such as fever, pain, inflammation, and other deviations from homeostasis. Non-steroidal anti-inflammatory drugs (NSAIDs) inhibit both COX enzymes. Non-specific (non-selective) NSAIDs block both COX1 and COX2. For example, aspirin, Ibuprofen, and Naproxen are common non-specific NSAIDs. Nonspecific NSAIDs are used for their anti-inflammatory, analgesic, anticoagulant, and antipyretic effects. However, they can produce unwanted side effects such as gastric bleeds, skin rashes, and bronchospasms (since some prostaglandins have a role in bronchodilation). In contrast, specific (selective) NSAIDs selectively inhibit COX2. Paracetamol (also called acetaminophen) is a common COX2-specific NSAID with potent analgesic and antipyretic effects. However, it does not have significant anti-inflammatory effects, and is toxic at high doses. Aspirin is unique to other NSAIDs because it primarily inhibits COX1 (though it also inhibits COX2). This has a significant impact on reducing clot formation, and is commonly used as a blood-thinner. Furthermore, aspirin modulates the functioning of COX2 enzymes such that they possess lipoxygenase activity, and actually produce anti-inflammatory lipoxin molecules. However, aspirin can lead to gastric ulceration, dyspepsia, nephritis, nausea, and hemorrhage. In the stomach, constitutive functioning of COX1 is important because prostaglandins inhibit gastric acid excretion. Without normal production of prostaglandins, there is a higher susceptibility to gastric ulcers. In the kidneys, normal activity of COX1 is important because prostaglandins are involved in the regulation of renal blood flow.
Also contributing to parturition are hormones from the fetus itself. Fetal stress stimulates the release of CRH from the fetus’ hypothalamus. Along with CRH from the placenta, CRH from the fetal hypothalamus drives ACTH release by the anterior pituitary, and consequent increases in cortisol. Fetal cortisol inhibits progesterone and estrogen secretion by the placenta and increases prostaglandin production by the placenta. Increased prostaglandins contribute to uterine contractions, but the decrease in progesterone is also important. Progesterone relaxes the smooth muscles of the myometrium, preventing their contraction. Cortisol has an antagonistic relationship with progesterone, and minimizes myometrial relaxation. This enables efficient contraction of the uterus during parturition.
 Nursing
Nursing
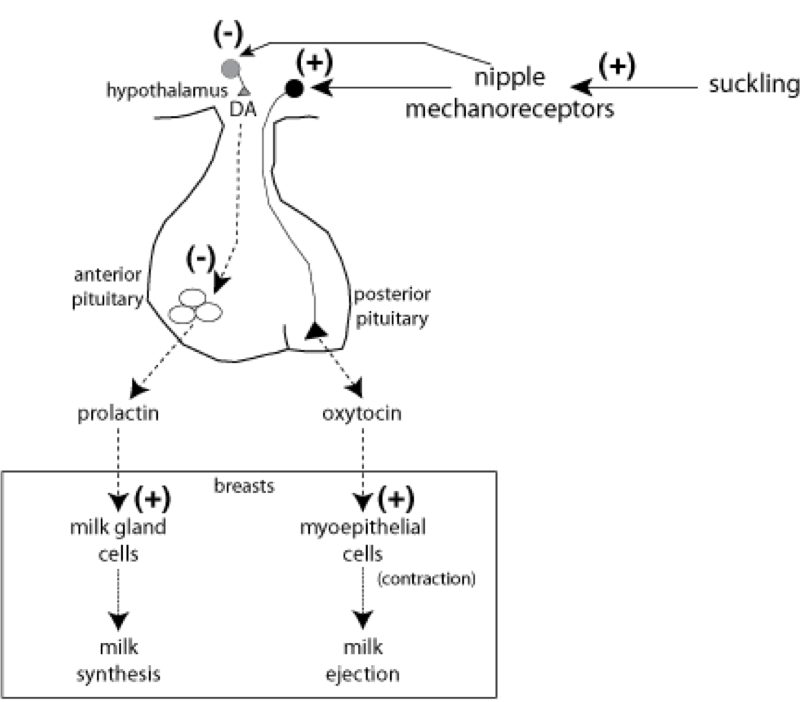
In addition to its role in parturition, oxytocin released from the posterior pituitary also plays a role in nursing. Suckling stimulates nipple mechanoceptors, which excite oxytocin-releasing neurons of the hypothalamus. Oxytocin stimulates contraction of the myoepithelium surrounding breast lobules that produce milk. Contraction of the myoepithelium causes excretion of the milk by the mammary glands. However, oxytocin does not alter milk production. Prolactin from the anterior pituitary stimulates milk production. In addition to stimulating oxytocin release, activation of nipple mechanoreceptor also inhibits dopaminergic neurons. Dopamine (among other signals) would normally travel through the portal system to the anterior pituitary and prevent the release of prolactin. So, by inhibiting such dopaminergic neurons, prolactin can be released.
TRH and vasoactive intestinal peptide (VIP) from the hypothalamus also drive the release of prolactin. In fact, many signals impact prolactin release. Lesions to the portal system between the hypothalamus and anterior pituitary can prevent stimulation of prolactin release by TRH and VIP. As mentioned, dopaminergic pathways can also inhibit prolactin release. As such, medications that reduce dopaminergic signaling (e.g., some antipsychotic and antidepressant treatments) can increase prolactin levels. On the other hand, certain growth hormones can lead to excessive prolactin release, as can estrogen, lending to the potential increase in prolactin following estrogen supplementation (e.g., from birth control). Lastly, hypothyroidism can even stimulate prolactin release due to elevated levels of TRH. Once released, prolactin can stimulate milk production by the mammary glands, and oxytocin can stimulate its excretion. Since suckling stimulates the release of oxytocin, suckling can also induce childbirth. The primary process through which prolactin is deactivated is through renal clearance, so any condition that leads to renal failure can also elevate prolactin levels.
Bonding
Oxytocin is also involved in bonding behavior (as such, nursing is involved in bonding behavior). This may have implications for formula-fed babies. However, oxytocin’s role in bonding is not limited to mother-child relationships. It also appears to have a role in partnership relationships, evidenced by the different mating behavior of prairie and montane voles. Montane voles of both sexes will mate with anything; they’re polyamorous. In contrast, prairie voles seem to mate for life. Female prairie vole bonding behavior is driven by oxytocin, whereas male prairie vole bonding behavior is driven by vasopressin (VP, also called antidiuretic hormone, ADH). Male prairie voles without functional vasopressin receptors exhibit polyamorous behavior comparable to montane voles, which lack the necessary vasopressin receptors. Arginine vasopressin (AVP) is the predominant type of vasopressin found in humans. Even though VP is only an 8-amino acid peptide, there is variation within humans in the types and ratios of VP released. Importantly, these findings in voles may not generalize to humans.
Puberty
In addition to the changes that can occur during embryonic development, puberty is also a time during which reproductive systems undergo dramatic physiological changes. These changes in reproductive systems also entail dramatic changes in other physiological systems. Puberty entails the development of primary sex traits, including enlarged gonads, enlarged ducts, enlarged sex glands, the production of sperm in males, and ovulation in females. During embryonic development, AMH is a masculinizing hormone only present in those destined to exhibit a male phenotype. In contrast, AMH increases throughout puberty in females, while decreasing in males. Puberty also entails changes in secondary sex characteristics, such as pubic hair growth. Additionally, puberty is a time of prefrontal cortex (PFC) maturation. While there is rapid PFC maturation in females, PFC maturation in males may take more than a decade after puberty onset. In females, psychosocial development has traditionally preceded or coincided with reproductive maturation, but following the industrial revolution, improvements in health and nutrition have advanced menarche, while increased psychosocial demands of modern society have delayed psychosocial maturation.
Precocious puberty is the early onset of puberty, though generally only the onset of secondary sex characteristic development, not necessarily of primary sex characteristics. There are multiple possible causes of precocious puberty, which can arise from central or peripheral origins. Central causes of precocious puberty include hypothalamic disorders and other CNS lesions, whereas peripheral causes can include premature adrenarche, congenital adrenal hyperplasia, steroid-secreting tumors, ovarian cysts, testoxicosis, gonadotropin-secreting tumors, and even hypothyroidism. Exogenous ingestion of steroids, such as a mother’s contraceptives or father’s testosterone replacement, may also precipitate precocious puberty.
Female Contraceptives
Hormone therapies can be used to alter the female menstrual cycle to limit the chance of pregnancy, generally by mimicking the hormonal environment of actual pregnancy. To achieve these desired effect, female hormonal contraceptives utilize the HPG negative feedback pathway to suppress GnRH and gonadotropin secretion, thereby preventing gonadotropins from stimulating follicle development that is otherwise necessary for ovulation. These mechanisms tend to revolve around the administration of progestins (synthetic progestogens), often combined with estrogens. Progestins stimulate only selective types of progesterone receptors (usually different from 17-hydroxyprogesterone), and reduce the likelihood of producing too many other steroid hormones (i.e., they do not serve as precursors for the production of other steroid hormones). Progestin administration inhibits the functioning of the endogenous progesterone cycle by mimicking the progesterone levels of pregnancy. It can eliminate the LH surge and cause a thin, atrophic endometrium. Progestins also thicken cervical mucus, and decrease the mobility of cilia in the fallopian tube. The latter, however, is concerning because if ovulation and fertilization do occur, there may be an increased risk of ectopic pregnancy; a failure to efficiently transport a fertilized oocyte to the uterus may increase the likelihood of implantation in a fallopian tube. Certain antibiotics can also decrease cilia mobility, and increase the risk of ectopic pregnancy. Estrogens suppress FSH and stabilize the endometrium (which helps control spotting). It also increases the efficacy of progestin by increasing intracellular expression of progesterone receptors. Taking progestin only can lead to low levels of estrogens through suppression of GnRH. Progestin only methods limit the risk of ovulation. However, by signaling pregnancy, progestin also has some adverse effects, such as mammary gland enlargement and tenderness, and increased food drive.
Daily oral contraceptives are the most common reversible form of hormonal contraception. “The Pill” is a combined oral contraceptive (COC); it includes both progestin and estrogen. This enables the pill to suppress ovulation. Other daily oral contraceptives include only progestin (e.g., the “minipill”). Different pills also have different durations during which active progestin and estrogen are consumed. For example, some include 3 weeks of active pills followed by one week off (or one week of placebo pills). Others include 12 weeks of active pills and one week off (or of placebo pills). The use of placebo pills, and extended use packs of pills, may increase efficacy through increased adherence. Up to 25% of females have a follicle prepared for ovulation by day 7 of the placebo week, so failure to maintain a regimen or failure to start a new pack increases the risk of ovulation.
There are also longer lasting hormonal administration methods, such as transdermal patches that incorporate a combined oral contraceptive into a 3-week patch followed by one week without the patch. This method has a failure rate of only 0.9%, and has higher compliance than daily contraceptive pills (88% vs. 78%). Other monthly methods also exist, including the vaginal ring (e.g., Nuva Ring). One ring is used each month, during which it is in the vagina for three weeks, and removed for one week. This provides constant low levels of hormones, and has a failure rate of only 1.2%.
Hormone injections are a very effective method, with a failure rate of only 0.3%. Depo-provera, for example, is a medroxyprogesterone acetate injection that inhibits ovulation and thickens cervical mucus and lasts for three to six months. Though extended use may risk bone loss and other problems associated with low estrogen, Depo-provera injections have several advantages. There are multiple durations, some of which can last up to 6 months. The long duration of action also enhances compliance. Medroxyprogesterone is effective for the extended duration because no enzymatic activity can alter it; it is only cleared in urine. Without estrogen, there is no increased risk of deep vein thrombosis, pulmonary embolism, stroke, or myocardial infarction. Depo-provera has minimal interactions with other drugs (especially compared to other hormonal contraceptives). There is an 80% reduction in the risk of endometrial cancer, as well as a decreased risk of iron-deficiency anemia, pelvic inflammatory disease, ectopic pregnancy, and uterine fibroids. It also decreases symptoms of endometriosis, decreases the incidence of primary dysmenorrhea, decreases ovulation pain, and decreases functional ovarian cysts, while also decreasing the incidence of seizures in females with epilepsy. Unlike other hormonal contraceptives, its effectiveness is not affected by anti-epilepsy drugs. Other methods are even longer-lasting, such as Implanon, an implanted subdermal progestin releasing rod that lasts 3-5 years, and inter-uterine-devices (IUDs), which can release progestin for up to 10 years.
Other methods still, often called emergency contraceptives, are only used episodically. Plan B (Levonorgestrel-only) is a massive dose of progesterone agonists that disrupts the uterine lining and prevents implantation when fertilization is suspected to have occurred.
GnRH agonists can also be taken to inhibit FSH and LH release. This simulates menopause, though the difference is that there is no increased production of FSH (and no consequent risk of ovarian cancer). Perpetually circulating GnRH agonists inhibit gonadotropin release by disrupting the pulsatile stimulation of receptors. Unless GnRH is released in a pulsatile fashion, it will fail to excite the anterior pituitary, and actually shuts down the pituitary. One example is Leuprorelin (branded as Lupron), which is sometimes used to reset menstrual cycles by temporarily inducing a pseudo-menopausal state. Additionally, GnRH agonists are also used to treat endometriosis.
Female contraceptive methods are not all hormonal. There are also permanent (but often reversible) contraceptive methods. These methods operate by preventing the passage of oocytes from the ovaries to the uterus through the fallopian tubes. Bilateral tubal ligation (BTL) involves cutting, sealing, clamping, or tying the fallopian tubes, thereby preventing the passage of oocytes. Bilateral tubal occlusion (e.g., Essure) involves blocking the fallopian tubes with a flexible insert, around which a natural barrier forms that prevents the passage of oocytes.
Male Contraceptives
Male hormonal contraceptive methods are less prevalent. However, they also primarily utilize the intrinsic negative feedback pathways of the HPG axis. The administration of exogenous androgens suppresses the pulsatile release of GnRH, LH, and FSH. By inhibiting LH, the endogenous production of gonadal androgens is suppressed. Under different circumstances, this would be problematic because it would result in low circulating androgens, but because the hormonal treatments themselves are androgen agonists, the diminished effects of endogenous androgens are, for the most part, replaced by the exogenous androgen agonists. More importantly than suppression of LH, androgen-mediated suppression of the HPG axis inhibits the release of FSH. Without FSH, spermatogenesis is impaired.
Gonadotropin suppression results in an acute decrease in type B spermatogonia. This prevents further differentiation of type B spermatogonia into primary spermatogonia, secondary spermatogonia, spermatids, and spermatozoa. However, the gonadotropin suppression does not have an acute effect on the development of spermatogonia that have progressed beyond type B spermatogonia. In other words, type B spermatogonia already present at the time treatment begins will be capable of developing into spermatozoa. This means that androgen treatments take a long time (at least several weeks) to induce spermatozoa concentrations low enough to serve as effective contraceptives.
Effective male hormonal contraceptives aim to achieve severe oligozoospermia, defined as concentrations of less than 1 million spermatozoa per milliliter. Achieving severe oligozoospermia translates into a failure rate of about 1% per year, comparable to the failure rates in the most efficacious female hormonal contraceptives. However, despite the dramatic decreases in spermatogenesis attributable to male hormonal contraceptives, there is a complete return of testicular function following cessation of contraceptive regimens.
Unfortunately, androgen-only male contraceptive treatments (such as testosterone enanthate, injected once weekly) have some significant limitations. Firstly, the doses of androgen-only contraceptives necessary to achieve severe oligozoospermia are about twice as high as normal physiological levels. This can lead to side effects associated with hyperandrogenemia, such as acne (the most common side effect) and male-pattern hair loss. Secondly, achieving severe oligozoospermia using androgen agonists takes about four months; they are not effective in the short term. Furthermore, about 10-15% of those treated never achieve severe oligozoospermia. The variability in responses to hormonal contraceptives is not attributable to differences in the degree of gonadotropin suppression, but it may result from differences in residual testicular activity, with some retaining sufficient androgenic activity to support spermatogenesis. There are also ethnic differences in male hormonal contraceptive efficacy, with Asian males being more responsive to hormonal contraceptive treatments than other ethnicities. However, the mechanisms underlying ethnic differences are unknown.
Another factor complicating male hormonal contraceptive treatments is the necessity to address changes in bone mineral density. Testosterone is very important for the maintenance of bone mineral density and as such, testosterone therapy can actually increase bone mineral density. However, bone mineral density depends, at least in part, on the aromatization of testosterone to estradiol; estradiol is important for male bone mineral density. Hormone therapies that shut down endogenous testosterone prevent the aromatization of testosterone to estradiol and may have a negative impact on bone mineral density. Estradiol may also be important for male sexual function and maintenance of body fat.
Certain contraceptive regimens have addressed some of the limitations of androgen-only male hormonal contraceptives. The degree of sperm suppression, and the rate at which that suppression is achieved, is enhanced by combining progestin with androgen agonists. Progestins also suppress gonadotropin levels, but their suppression of gonadotropin levels is additive to the suppressive effects of exogenous androgens. As a result, lower androgen doses (i.e., normal physiological doses of androgens) are sufficient to achieve the same degree of gonadotropin suppression when combined with progestins. This minimizes the risks associated with the high doses of androgens necessary to achieve oligozoospermia when administered as the sole contraceptive treatment. Furthermore, unlike androgen agonists, which primarily reduce spermatogenesis through suppression of FSH, progestins may also have direct effects on testicular function that inhibit spermatogenesis.
One male contraceptive regimen incorporating a progestin agonist is testosterone combined with nestrone. Nestrone is a progestin derived from 19-norprogesterone. Nestrone has no androgenic, estrogenic, or glucocorticoid activity, which makes it less likely to produce the side effects attributable to some other progestins. The combination of testosterone and nestrone enables 89% of men to achieve severe oligozoospermia. Furthermore, because testosterone is aromatizable to estradiol, the regimen does not pose significant risks to bone mineral density.
Another emerging treatment is dimethandrolone undeconoate (DMAU), a synthetic 19-norandrogen. DMAU can be taken either orally, or injected. In vivo, DMAU is hydrolyzed into dimethandrolone (DMA), which is a potent agonist at both androgen and progesterone receptors. This makes DMAU a viable single-agent contraceptive. However, DMA cannot be aromatized to estrogen, posing potential risks to bone mineral density.
One other potential treatment is 17-α-methyl-19-nortestosterone (MENT), a highly potent synthetic androgen (more potent than testosterone). MENT is formulated as an implant for long-term hormonal contraception, with sustained MENT release over the course of a year. MENT is resistant to 5α-reduction, which may contribute to long-term prostate health by reducing androgenic activity in the prostate. MENT is also aromatizable, so it probably would not have a negative impact on bone mineral density. However, MENT does not have progesterogenic activity.
In addition to the easily reversible male hormonal contraceptives, there are also non-hormonal contraceptive methods, mainly vasectomy. Vasectomy involves severing the vas deferens, thereby preventing the inclusion of spermatozoa in the ejaculate. While this procedure has historically resulted in permanent sterilization, advances in surgical technologies and techniques now permit a high success rate for vasectomy reversal.
Other Steroid Hormone Treatments
Steroids can also be taken for other purposes. For example, anabolic steroids are often used as doping compounds for physical enhancement. However, such compounds are often converted to other active hormones, which can lead to unintended downstream effects. Methyltestosterone is a testosterone agonist that isn’t broken down as easily as testosterone. However, it is readily converted to estrogen, leading to increased stimulation of estrogen receptors. High amounts of estrogen in females suppresses FSH, reducing the likelihood of ovulation, but in males, high estrogen reduces sperm count, and reduces local paracrine effects of testosterone production. It can also lead to testicular reduction. Fluoxyltestosterone is another testosterone agonist, but it is not converted to estrogen. Rather, it is converted to cortisol. This can lead to “roid rage.”
Sexual Spectrum
The reproductive physiology discussed thus far has been generalized for males and females. In reality, there is much greater variation in the reproductive physiology because there not just two discrete categories of sex. In fact, there is a spectrum of sexes between the stereotypical XY male and the stereotypical XX female, and different aspects of sex can be classified in different ways. For example, though a person may have XY chromosomes and the associated primary sexual characteristics (i.e., testes, etc.), a male may develop enlarged mammary glands, a disorder called gynecomastia. This can occur when the balance between estrogen and testosterone is disrupted, and can happen via several physiological or non-physiological mechanisms. Physiological gynecomastia has a trimodal age distribution. For example(s), it may be a neonatal consequence of the uterine environment. It may also occur during adolescence due to a 5α-reductase deficiency (preventing the ability to produce 5α-DHT). Lastly, it can occur in adults due to Leydig or Sertoli cell tumors, or due to certain drugs (e.g., diazepam, Prilosec, THC). Gynecomastia creates a disconnect between primary male sexual characteristics and secondary female sexual characteristic. But this is only the tip of the iceberg.
There is a distinction between chromosomal sex, gonadal sex, phenotypic sex, and sexual identity. Though the ‘norm’ is for an alignment of all of these aspects of sex as either feminine or masculine, there are numerous and relatively common incidences of dissonance. In other words, instances of dissonance between chromosomal sex, gonadal sex, phenotypic sex, and sexual identity are not necessarily ‘abnormal.’ For the purpose of comparison, though, we will refer to those with chromosomal sex, gonadal sex, phenotypic sex, and sexual identity in alignment as ‘normal.’
Chromosomal sex refers to the presence of XX, XY or some other chromosomal variation. Klinefelter’s Syndrome (KS), is the presence of XXY (47 total chromosomes). The phenotype is not substantially different from a normal XY male phenotype. The changes have to do with the presence of the X chromosome suppressing some of the Y chromosome genes. Those afflicted are almost always sterile, though there is still a chance of having children through assisted reproductive technologies. They may also have weaker muscles, greater height, poor coordination, less body hair, smaller genitals, breast growth (gynecomastia only present in about 1/3 of those affected, usually not to a degree that would prompt cosmetic surgery), and less interest in sex. Symptoms often emerge during puberty. Reading difficulties and problems with speech are common, though intelligence is usually normal. Symptoms are more severe if three or more X chromosomes are present. There is also a higher risk of breast cancer than normal XY males, but still lower than XX females. Nevertheless, there is a normal life expectancy. The disorder is also relatively prevalent: between 1/500 and 1/1000 males are affected. There is often low serum testosterone, and high serum FSH and LH.
In contrast to the extra X chromosome in Klinefelter’s Syndrome, Turner Syndrome (TS) is the partial or complete absence of a second X chromosome (X0; 45 chromosomes). A single functional X chromosome is not sufficient for expression of certain traits; two are necessary for normal expression of some genes. Turner syndrome can result in a webbed neck, low ears, low hairline on the back of the neck, short stature, swollen extremities, absence of breasts and menstruation (when not treated with hormone therapy), heart defects, diabetes, low thyroid hormone levels, and visual/auditory problems. Most of those afflicted have normal intelligence, but may have trouble with spatial visualization. Turner syndrome may be treated by increasing the expression of receptors, supplementing growth hormones to increase height, and supplementing estrogen to promote secondary sex characteristics.
Gonadal Sex is the presence of testes versus ovaries, which determines which hormones are produced in the gonads. As mentioned, this is determined in utero by the transcription of SRY genes in males, the products of which induce differentiation of bipotential gonads into testes. Without the SRY genes found on the Y chromosome, the gonads differentiate into ovaries.
Phenotypic sex refers to the presence of a masculine versus feminine appearance. Congenital adrenal hyperplasia (CAH) results from mutations in the genes for enzymes that mediate corticosteroid synthesis pathways. However, CAH can entail an upregulation or downregulation of sex steroids. When CAH results in excessive androgen production, there may be ambiguous genitalia in females, namely clitoromegaly, enlarged clitoris, and shallow vagina. There may also be early pubic hair development and rapid growth in childhood. This can result in precocious puberty or failure to undergo puberty. There may also be excessive facial hair, virilization, or menstrual irregularity in adolescence. Infertility is common due to anovulation. When CAH results in insufficient levels of adrenal androgens, there is under-virilization in XY males, which can result in apparently female external genitalia. In females, hypogonadism can cause sexual infantilism or abnormal pubertal development, infertility, and other reproductive system abnormalities. The most common type (95%) of CAH involves mutations in the gene for 21-hydroxylase that leads to 21-hydroxylase deficiencies. This leads to an overproduction of reproductive hormones, and an underproduction of glucocorticoids and mineral corticoids. This can be identified by hypoglycemia (due to hypocortisolism), hyponatremia (due to hypoaldosteronism), hyperkalemia (due to hypoaldosteronism), and elevated 17α-hydroxy-progesterone. Males with classic CAH generally have no symptoms at birth, though some may be hyperpigmented due to elevated MSH secretion. Notably, the hyperplasia in classic CAH results in a lack of feedback to the hypothalamus and anterior pituitary by glucocorticoids. This leads to elevated levels of CRH and ACTH. The elevated ACTH drives hyperplasia of the adrenal cortex, but is unable to sufficiently induce glucocorticoid production due to the impaired activity of 21-hydroxylase. This is comparable to instances of hypothyroidism that result in a goiter, in which the failure to adequately produce thyroid hormones in response to TSH leads to a lack of negative feedback, and a consequent increase in TSH that drives hyperplasia of the thyroid, yielding a goiter.
Androgen Insensitivity syndrome (AIS) is the presence of XY chromosomal sex, but the absence of functional androgen receptors during developmental stages, which leads to a hyper-feminization of secondary sex characteristics while retaining internal male genitalia. This can be due to mutated receptors with lower affinities, the absence of receptor genes, a failure to express receptor genes, or a failure to express receptor genes in the right places at the right time. Though the result is female external genitalia and non-mature ovaries, AIS is still a spectrum due to the diversity of causes; some cases are more severe than others.
Sexual identity encompasses masculine versus feminine identity and sexual orientation. Keep in mind that there can be significant variation in sexual identity between individuals, and within a single individual over the course of their lifetime. Gender is more of a cultural construct. Bruce Jenner’s transition to Caitlyn Jenner is an example of how sexual identity can change over the course of a lifetime. It also demonstrates how secondary sex characteristics are dependent upon sex steroids (e.g., estrogens in the case of Jenner). Males have estrogen receptors, but they don’t produce the same types of estrogens or in the same quantities. Masculinized humans that want to feminize only need to take estrogens. Feminized humans that want to masculinize need to take lots of testosterone, and it still won’t work well due to the low expression of testosterone receptors. In fact, females normally have circulating androgens, but do not have a pattern of receptor expression that allows androgens to have dramatic masculinizing effects. In fact, adrenal androgens in females are important for normal libido. Taking 5α-dihydrotestosterone (5α-DHT) will start to masculinize a feminine human, but 5α-DHT also increases the risk of cardiac complications. As females age, they produce less estrogen, and their androgen levels increase. The rise in endogenous androgens drives the production of androgen receptors. This causes females to begin exhibiting secondary masculine characteristics, such as facial hair, deeper voices, etc.
5α-DHT is of vital importance in males. As mentioned previously, it binds with 2-3 times stronger affinity than testosterone, but it also has a longer half-life (53 minutes as opposed to 34 minutes). It is particularly important for both embryonic and adolescent development of male characteristics. Unlike other androgens, 5α-DHT cannot be converted to estrogens. However, increased production of estrogens in males (e.g., due to increased aromatase expression) removes the precursors necessary to produce 5α-DHT and can lead to deficits in development of male sex characteristics.
Feedback/Errata