
Muscle Physiology
Charles Asbury
Introduction: Why is muscle physiology important?
You may see patients with muscle-related diseases and you may prescribe treatments that act directly on muscles. Muscle physiology can also have a major impact on surgery and interventions that are intended to treat other organs. In addition, a wide variety of medical problems, including ionic disturbances, exposure to toxins or drugs, and congenital disorders can lead to detectable changes in muscle function, which therefore serve as diagnostic clues. Understanding how electrical excitation of muscle leads to its contraction will also help you learn about neurophysiology, signal transduction pathways, and motor proteins.
Session Learning Objectives
- Explain the mechanism by which muscle contracts, outlining how the sliding of actin filaments in sarcomeres is driven by ATP-dependent chemo-mechanical cycling of myosin motor proteins.
- Explain excitation-contraction coupling and relaxation in skeletal muscle by identifying the roles of the t-tubules, calcium channels (Cav1.1 and the ryanodine receptor), thin filament regulators (troponin and tropomyosin), and ATP-dependent calcium pumps.
- Compare twitch contractions for slow/type 1 and fast/type 2 skeletal muscle fibers and explain the molecular bases for the differences in twitch behavior. Define isometric and isotonic contractions.
- Explain how smooth, graded contractions of a skeletal muscle are produced by changes in stimulus intensity and by the size principle of motor unit recruitment.
- Understand the differences in excitation-contraction coupling between skeletal, cardiac, and smooth muscle. Describe the two-stage phospho-regulatory cascade that initiates smooth muscle contraction.
- Compare and contrast how skeletal, cardiac and smooth muscle are controlled by the nervous system. Define single-unit vs. multi-unit smooth muscle types.
Muscle generates force by contracting against external load. There are three main types of muscle, skeletal, cardiac, and smooth. All three use the same fuel source, ATP, which supplies chemical energy that muscles convert into mechanical work and heat. All three share the same fundamental mechanism for generating force. However, they vary in their morphology, physiology, biochemical properties, and mechanics. Here you will begin to distinguish similarities and differences in how they function (in their physiology). We will start by reviewing skeletal muscle.
Note about muscle nomenclature: Some familiar cell structures are given muscle-cell-specific names. It will be useful to know these terms:
- sarcolemma = muscle outer (plasma) membrane (sarco = Greek for “flesh”; lemma = “husk”)
- sarcoplasm = muscle cytoplasm
- sarcoplasmic reticulum = specialized endoplasmic reticulum in muscle; stores Ca2+
The contraction machinery in skeletal muscle is organized hierarchically
Recall from your prior histology sessions that skeletal muscles are organized in a hierarchical manner, from the whole organ down to the individual molecules that drive its contraction (see Figure 1 and notes from Muscle Histology). Their organization gives them a striated appearance in the light and electron microscopes. It is also important for their function.
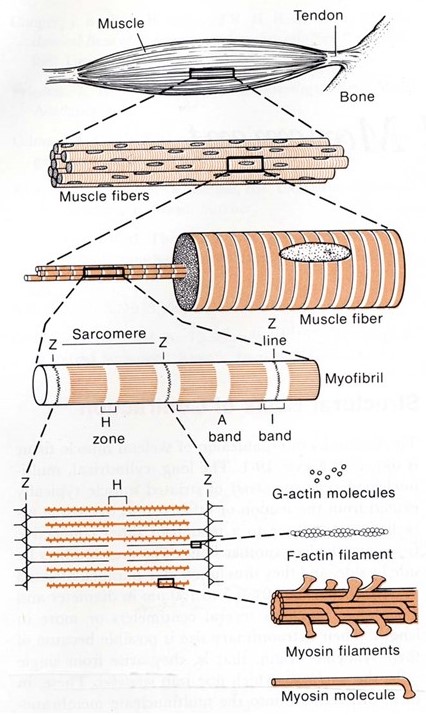
Each whole muscle is composed of hundreds of muscle fibers, roughly ten to twenty micrometers in diameter. Each fiber is a single multi-nucleated cell, surrounded by a plasma membrane (for muscle cells this is often given a special name, the sarcolemma) and containing nuclei, mitochondria, and other organelles common to all living cells. Every muscle fiber contains hundreds of myofibrils, each about a micrometer in diameter. Every myofibril is a series of sarcomeres, linked end-to-end down the entire muscle fiber. Inside each sarcomere is a set of hundreds of parallel thick and thin filaments (Figure 2). The thin filaments extend into the sarcomere from the left and right sides. The thick filaments extend outward from the centerline, and these contain the primary force-generating molecules, called myosin.
Muscle shortening is driven by relative sliding of thick and thin filaments
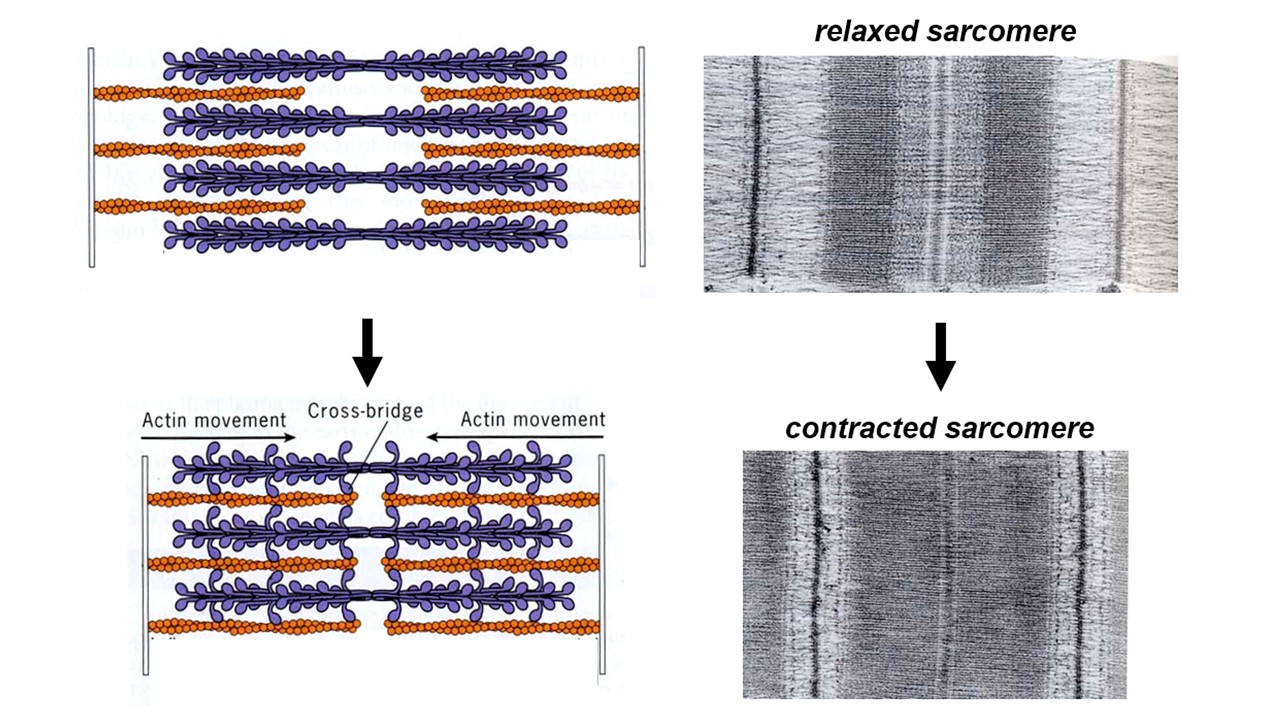
Normally the thin and thick filaments overlap. When a muscle cell contracts, the amount of overlap between thick and thin filaments increases and the thick filaments in one sarcomere move closer to those in the next sarcomere of the series (i.e., the dense striations move closer together, as can be seen in the electron microscope – see Figure 2). Muscle contraction is driven by interactions between the thick and thin filaments that drive sliding of the two sets of filaments relative to one another. The thick filaments literally crawl along the thin filaments, and that crawling generates active force.
High-resolution electron micrographs show tiny projections from the thick filaments that often bridge across and attach to an adjacent thin filament. In some images, these cross-bridges appear to tilt all in the same direction (particularly when the muscle is in “rigor“, i.e. lacking ATP and therefore tightly locked up). Tilted cross-bridges led Hugh Huxley to propose (in 1969) that they drive filament sliding by cyclically attaching to a thin filament, tilting, detaching, and then un-tilting. The tilting of cross-bridges around a hinge point within the myosin head is now well-proven.
Key molecules of the sarcomere
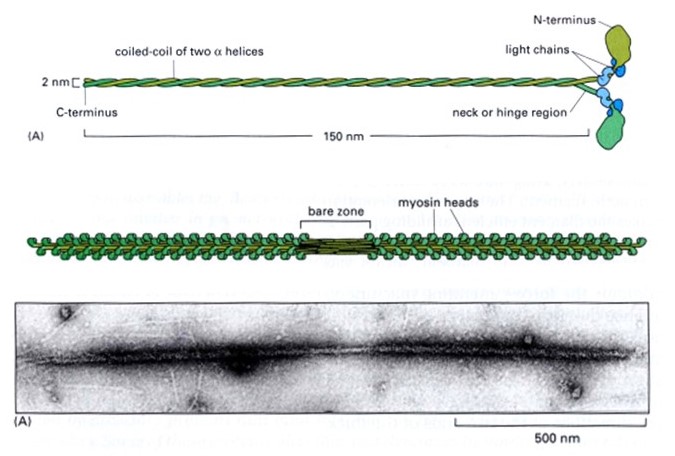
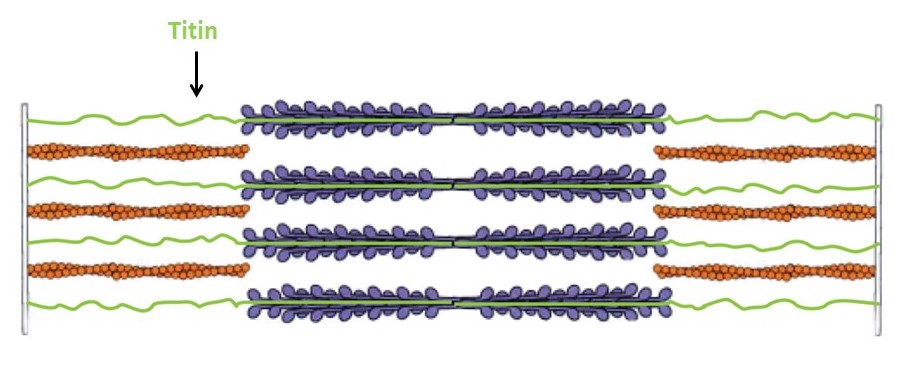
The thick filaments are composed primarily of the protein myosin (Figure 3). Myosin is made of three protein chains that form a six-member, rod-like complex (two copies of each chain). The largest protein chain (sometimes called the ‘heavy chain’) forms a long coiled-coil with twin globular ‘head’ domains at one end. Smaller protein chains (‘light chains’) bind at the junction between the coiled-coil rod and the heads (these form the ‘lever arm’, that rotates, as described below). To form one thick filament, several hundred myosin molecules assemble in a bipolar fashion, with their coiled-coil rods packed together in a bundle, and with their globular heads sticking out from the surface of the bundle at regular intervals. These heads sticking out from the bundle are the cross-bridges that drive sarcomere shortening by cyclic interactions with the thin filaments. Another element of the thick filament is titin (Figure 4). Titin is a very long and elastic protein that binds along each thick filament and extends beyond the tip of each thick filament to the side/end of the sarcomere (to the Z-discs). Even when a muscle is relaxed, titin provides it with passive elasticity, and titin also keeps the thick and thin filaments in register with one another, especially when their degree of overlap is low during a strong muscle stretch.
The thin filaments are composed primarily of actin. Actin is a “globular” (i.e., roughly spherical) protein that self-assembles into a helical polymer. The globular form is sometimes called G-actin, and the polymeric form, F-actin (F for “filamentous”). To form F-actin, hundreds of G-actin subunits pack together into two rows that gently wrap around one another, in a rope-like arrangement. Thin filaments also contain some additional elements besides actin. Of these, tropomyosin and troponin are the most important. Tropomyosin is a rod-like protein that binds along the groove of F-actin. Troponin is a globular protein that binds periodically on the thin filament, interacting with both the actin and the tropomyosin. Tropomyosin and troponin regulate skeletal muscle contraction, and we will discuss them in some detail in the section on excitation contraction coupling, below.
Atomic-level structures of myosin fragments suggest a detailed picture of myosin’s cyclical action (Figure 5). Each myosin head contains a lever-arm between the actin-binding portion and the coiled-coil rod. The lever arm is found in a very different orientation in ATP- versus ADP-bound structures, suggesting that it undergoes a large rotation during the myosin cycle. Biochemical experiments show that myosin binds more tightly to actin in the absence of nucleotide (or in the presence of ADP) than with ATP. These observations together with the atomic structures provide strong evidence supporting the tilting cross-bridge model for muscle contraction.
Chemo-mechanical cycle of myosin
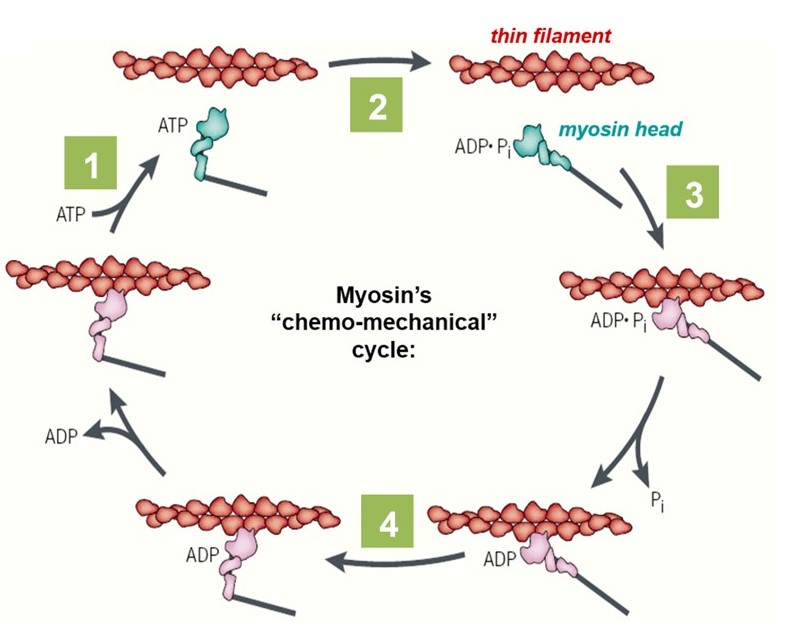
The lever-arm motion of a myosin head (cross-bridge) is tightly coupled to the hydrolysis of ATP, through its “chemo-mechanical cycle” (Figure 5). The nucleotide (i.e., ATP or ADP) participates in several key transitions in the cycle: First, (1) ATP binding to a myosin head triggers detachment of the myosin from the actin filament. Next, (2) hydrolysis of the bound ATP converts the head into a pre-stroke state, in which its lever arm adopts a mechanically strained configuration. It is literally ‘cocked’ into a high energy state, similar to the firing pin of a gun. A myosin head in the pre-stroke state has a higher affinity for actin. (3) Attachment of the head to actin triggers release of ADP and phosphate, which in turn (4) triggers the ‘power stroke’, when the lever arm rotates back to its low energy configuration. This power stroke drives movement of the actin filament and generates force. The chemo-mechanical cycle of myosin is diagrammed in Figure 5 and animated in the 2-minute video below (includes narration).
Biophysics side note: How much force does an individual myosin molecule need to generate to produce the strength of a typical muscle? To estimate this unitary force, the number of myosins acting together in parallel must be estimated. Cross-sections through skeletal muscle viewed by electron microscopy show ~400 thick filaments per square µm. A muscle 5 cm in diameter would therefore have 800 billion (800×109) thick filaments acting in parallel (i.e., 2.5×104 µm squared times π, times 400 thick filaments per square µm). Electron micrographs of isolated thick filaments suggest ~300 myosin heads emanating from each thick filament (e.g., see Figure 3). Multiplying these two numbers gives 2×1014 myosin heads acting in parallel over the cross section of the entire muscle (i.e., 800×109 thick filaments times 300 myosins per thick filament). This is an amazingly large number!!! Skeletal muscles have evolved such an orderly structure that 1014 molecules can all be tugging simultaneously on the same load. That’s 10,000 times more molecules cooperating within a single muscle than there are people on earth! Dividing a 50 lbs (225 Newtons) force by this huge number of myosins gives about 1×10-12 N, or 1 pN per myosin. (There is no need to memorize these numbers. Just appreciate their magnitude).
Using laser traps (a.k.a. ‘optical tweezers’), it is possible to directly measure the motion and force produced when a single myosin molecule binds an actin filament and undergoes one lever arm rotation (one ‘power stroke’). A single myosin from skeletal muscle can generate ~10 nm of movement against ~3 pN of force, more than enough to explain muscle contraction given the massive numbers of molecules involved.
Simple exercises to check what you recall
The job of skeletal muscle is to contract when commanded by the nervous system. The command comes in the form of synaptic input from a motor neuron, which impinges on the muscle cell at the neuromuscular junction. Here we will cover how this input triggers muscle contraction, a process called excitation-contraction (EC) coupling. We will begin by focusing on EC coupling in skeletal muscle. This will serve as a foundation for our discussion below about EC coupling in cardiac and smooth muscle.
The mechanism of excitation-contraction coupling in skeletal muscle can be summarized in six steps, listed here and shown schematically in Figure 6.
- Somatic motor neuron releases acetylcholine (ACh) at neuromuscular junction.
- Entry of Na+ through nicotinic ACh receptor (nAChR) channels initiates a muscle action potential.
- Action potential travels deep into muscle fiber along t-tubules.
- Voltage change in t-tubules triggers conformational change in Ca2+ channels (Cav1.1 and ryanodine receptor [RyR]) to release Ca2+ from internal stores.
- Ca2+ binding to regulatory proteins (troponin and tropomyosin) exposes myosin binding sites on actin, allowing contraction to begin
- Ca2+ is pumped into internal stores or extracellularly to stop contraction.
Each step is explained in more detail in the descriptions that follow.
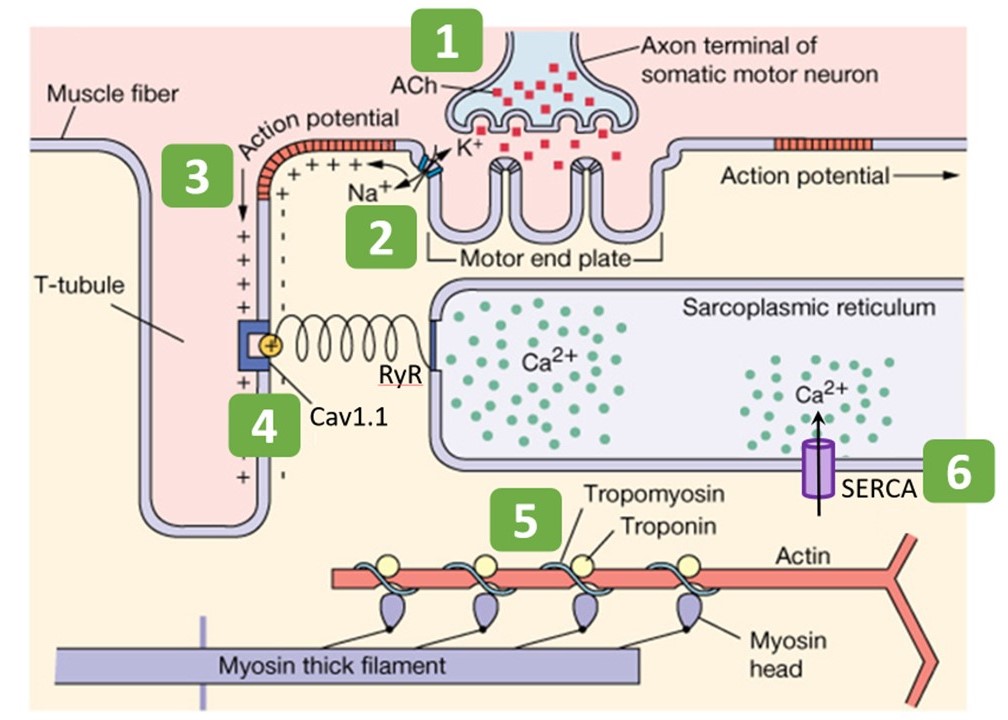
Step 1: Somatic motor neuron releases acetylcholine at neuromuscular junction
You have already learned in previous sessions on action potentials and synapses the basic steps for a neuron to release neurotransmitter. Skeletal muscle receives synaptic input from a somatic alpha motor neuron at the neuromuscular junction. This is the initial step in excitation. In this case the neurotransmitter is acetylcholine (ACh). Muscle contraction relies on the binding of acetylcholine to postsynaptic nicotinic acetylcholine receptors (nAChRs) at the motor end plate (Figure 6). Acetylcholine that does not get immediately degraded by acetylcholinesterase in the synaptic cleft binds to nAChRs. (Acetylcholinesterase is omitted from Figure 6.)
Pharmacology note: nAChRs are so named because nicotine is an agonist for these receptors. Do not confuse these with muscarinic acetylcholine receptors (mAChRs), which can be bound by muscarine, and are involved in smooth and cardiac muscle synaptic transmission (as described in sessions on autonomic nervous system). Note also the two different signal transduction schemes: nAChRs are fast ligand-gated ion channels, whereas mAChRs are G-protein coupled receptors (not ion channels).
Step 2: Net entry of Na+ through receptor channel initiates a muscle action potential
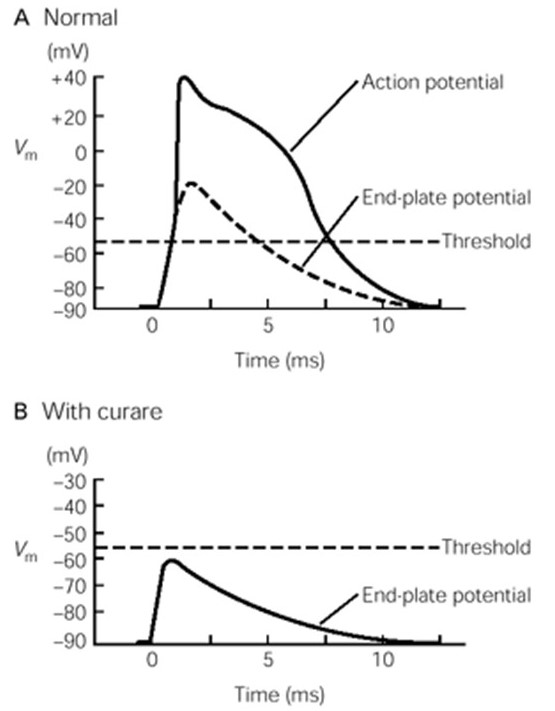
nAChRs are direct ligand-gated cation channels. Their activation leads to depolarization of a portion of the sarcolemma. The depolarization is called an end-plate potential, or EPP (Fig. 9). nAChRs are permeable to both Na+ and K+ (Figure 6), which move through the channels according to their electrochemical gradients (as explained in the prior session on membrane electricity). Since Na+ and K+ have roughly equal concentration gradients in opposite directions, the membrane potential of the muscle goes to about halfway between ENa and EK, around 0 mV, assuming equal permeability to both ions. (To review your understanding of membrane electricity, consider what would happen to the EPP if the permeability of the nAChR for Na+ were greater than for K+?) The combined influx of Na+ and efflux of K+ is the end-plate current. (How would a change in extracellular ion concentrations alter the end-plate current?)
The end-plate potential is a graded depolarization – its amplitude will vary with the number of receptors bound by ACh. Typically, however, the end plate potential in a muscle cell is always large enough to reach the threshold for activating the voltage-gated Na+ channels along the sarcolemma that will elicit a muscle action potential, the beginning of the excitation / contraction coupling of the muscle fiber.
Step 3 : Action potential travels deep into the fiber along t-tubules
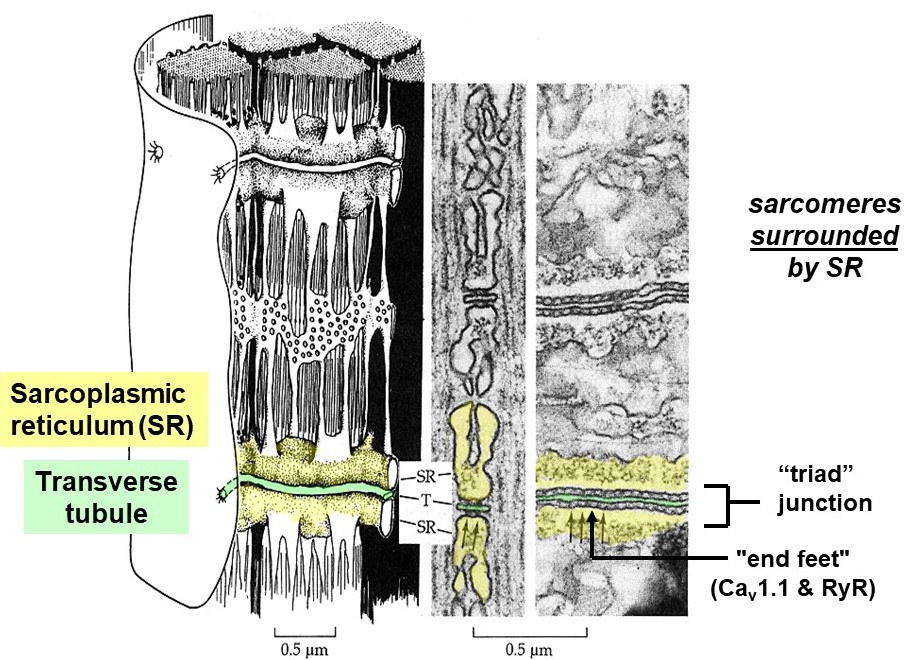
The action potential propagates deep into the muscle cell along t-tubules, membranous tubes that are continuous with the sarcolemma. The t-tubule network is akin to a system of telegraph wires that deliver the message, “contract!”, very quickly and nearly simultaneously to all the contractile machinery inside the muscle cell. To do so, the t-tubules pass very close to another membranous compartment inside the cell, the sarcoplasmic reticulum (SR), which stores lots of calcium. The junction where t-tubules and SR pass very close together, when viewed by electron microscopy, contains regularly spaced, dense particles bridging between the two membranes (Figure 8). These particles, sometimes called “end feet”, contain two key membrane-embedded proteins, Cav1.1 and the ryanodine receptor (RyR). (Note that Cav1.1 is also known as the dihydropyridine or DHP receptor, and also as the L-type calcium channel.)
Step 4: Voltage change in t-tubules causes conformational change in channels to release calcium from internal stores
The Cav1.1 proteins are voltage-sensitive calcium channels embedded in the t-tubule membranes. When an action potential arrives at the t-tubule, Cav1.1 channels open and allow calcium to flow from the lumen of the t-tubule (which is continuous with the exterior of the cell) into the muscle cell sarcoplasm (Figure 8). However, the amount of calcium entering the muscle cell from the exterior via Cav1.1 channels is low compared to what is needed for contraction. A more substantial, internal source of calcium is required, called the sarcoplasmic reticulum (SR). The SR is a system of internal membrane-bound calcium storage compartments surrounding each sarcomere and located very nearby each t-tubule. In skeletal muscle, the t-tubule Cav1.1 channels are bound directly to another class of calcium release channels embedded in the SR membrane, called ryanodine receptors (RyRs). This direct, mechanical coupling of t-tubule Cav1.1 channels to RyRs in the SR allows the two channels to cooperate and quickly flood the muscle cell with calcium. Voltage-dependent conformational changes in Cav1.1 directly push the RyRs open, releasing a flood of calcium from within the SR into the muscle cell sarcoplasm.
Step 5: Calcium binding to regulatory proteins exposes myosin binding sites on thin filaments, allowing contraction to begin
In resting muscle, actin and myosin do not interact because tropomyosin on the thin filaments obstructs their binding. Calcium released into the muscle sarcoplasm binds to the regulatory protein, troponin, located on the thin filaments. Calcium binding to troponin causes a conformational change that moves tropomyosin out of the myosin-binding groove on actin (Figure 9). Exposure of the myosin-binding groove on actin enables chemo-mechanical cycling of myosin to generate contractile force (see Figure 5 and Learning Objective #1 above for details).
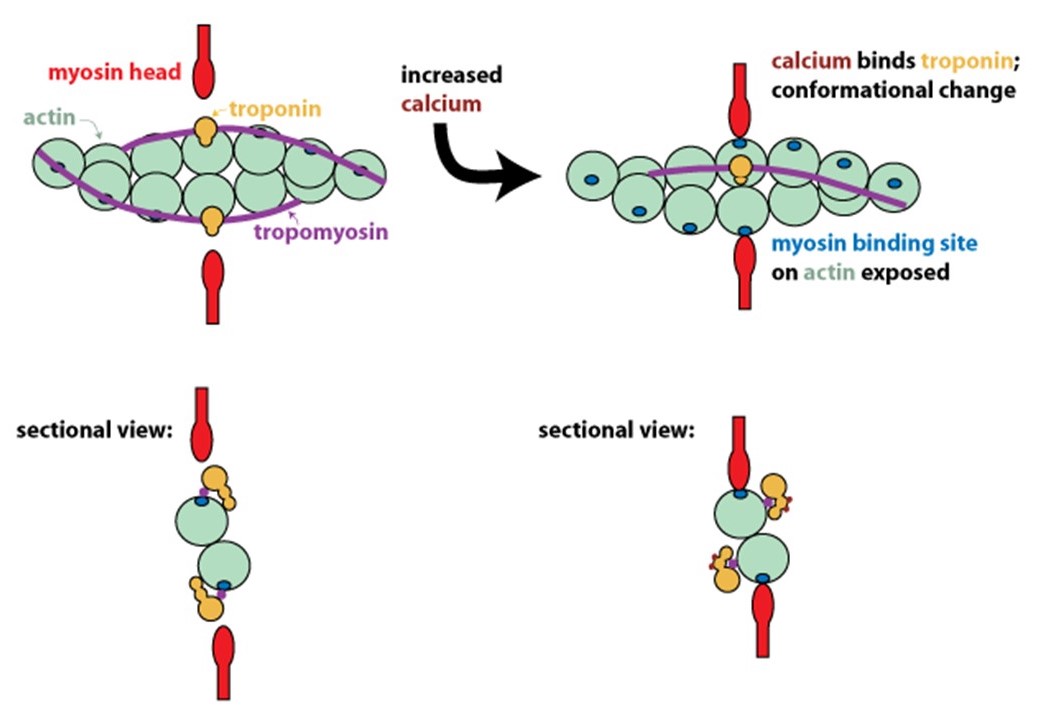
Step 6: To stop contraction, calcium is pumped back into internal stores and extracellularly
Because calcium is the trigger for muscle contraction, its presence in the sarcoplasm is highly regulated. To stop contraction, calcium ions are sequestered into the sarcoplasmic reticulum by membrane transport proteins called sarco/endoplasmic reticulum calcium ATPases (SERCAs). Sodium-calcium exchangers also pump calcium out of the sarcoplasm into the extracellular space through secondary active transport (not shown in Figure 6).
Skeletal muscle relaxation also requires that the alpha motor neuron stops firing action potentials. ACh is continuously removed from the synaptic cleft by acetylcholinesterase (AChE), thereby terminating the input signal to the muscle. Cross-bridge cycling will cease when Cav1.1 and RyR channels close, and when SERCAs on the SR pump the calcium out of the sarcoplasm. As calcium levels in the sarcoplasm fall, troponin releases calcium and allows tropomyosin to slide back into place over the myosin-binding sites. Actin and myosin are then prevented from interacting. Relaxation can occur quickly but is not instantaneous, because it takes some time to sequester the calcium. Chloride ions (Cl–) are also involved. Chloride enters the skeletal muscle cell during relaxation, when voltage-gated CLC-1 channels open. The influx of chloride ions speeds the repolarization of the muscle cell’s membrane potential, thereby accelerating the relaxation process.
Pharmacology note: Acetylcholinesterase (AChE) inhibitors are chemicals that inhibit the AChE enzyme from breaking down ACh, thereby increasing the level and prolonging the action of the neurotransmitter. These pharmacological agents are used to assist people with certain neuromuscular junction disorders, but they can lead to side effects in the autonomic nervous system.
Simple exercises to check what you recall
As mentioned above, a single action potential in a skeletal muscle cell nearly always releases a maximal amount of calcium from the SR, producing a twitch contraction of nearly uniform strength in every instance. The action potential itself is fast, lasting only a couple of milliseconds, and calcium floods the sarcomeres after only a short delay. Peak (maximum) tension may take a few milliseconds to develop, or a few tens of milliseconds, depending on the fiber type. The rate at which force develops mainly reflects the rate at which calcium binds troponin, moves tropomyosin, and gradually allows engagement of myosin motors with thin filaments. If the electrical stimulation then ceases, the tension falls back to zero in about 50 to 500 ms, again depending on the fiber type. The rate of relaxation reflects the rates at which myosin releases F-actin, and the rate of re-sequestration of calcium.
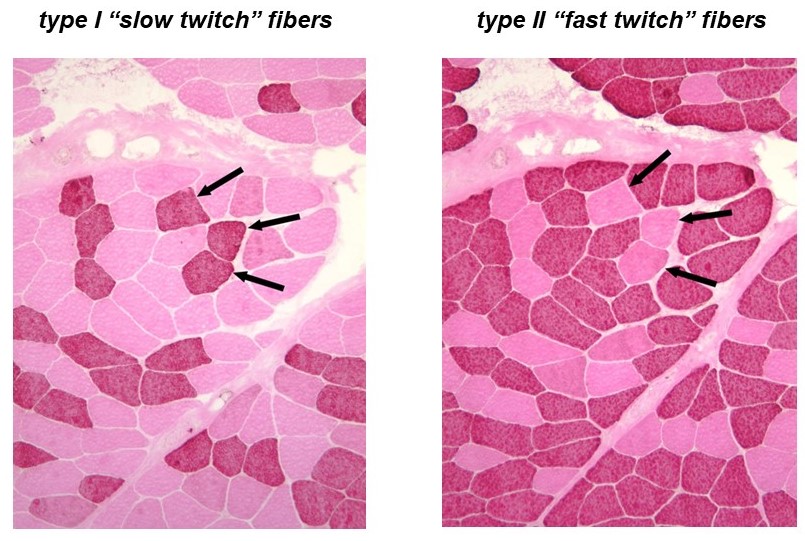
Recall that a whole skeletal muscle consists of many individual muscle fibers, each of which is a single, multinucleated cell (Figure 1). Whole skeletal muscles are mosaics, consisting of mixtures of ‘slow twitch’ (type I) and ‘fast twitch’ (type II) fibers (Figure 10). In response to a single action potential, type I slow twitch fibers develop force more slowly and they also relax more slowly (Figure 11). Their slower development of force is partly explained by a lower density of RyR and Cav1.1 channels, which implies that calcium will be released more slowly from their SR. Their slow relaxation is partly explained by a lower density of SERCA pumps, which implies that calcium will be sequestered into their SR more slowly. The different fiber types also have different isoforms of myosin, with different ATPase rates, and therefore different intrinsic speeds. Intrinsic speeds for the various myosins, measured in gliding filament assays correlate well with shortening speeds of the fiber types in which they are found (Figure 12). (Note: Gliding filament assays were explained in the prior session on Cytoskeleton & Motor Proteins.) This correspondence suggests that ‘slow twitch’ fibers are slow in part because the myosin isoform they contain has an intrinsically slower chemo-mechanical cycling rate.
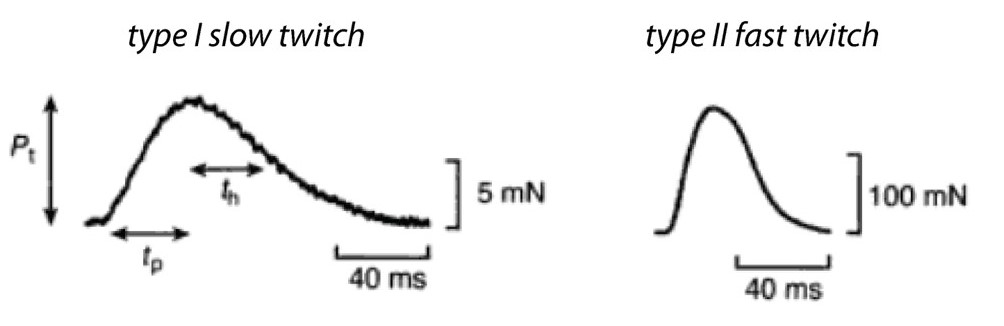
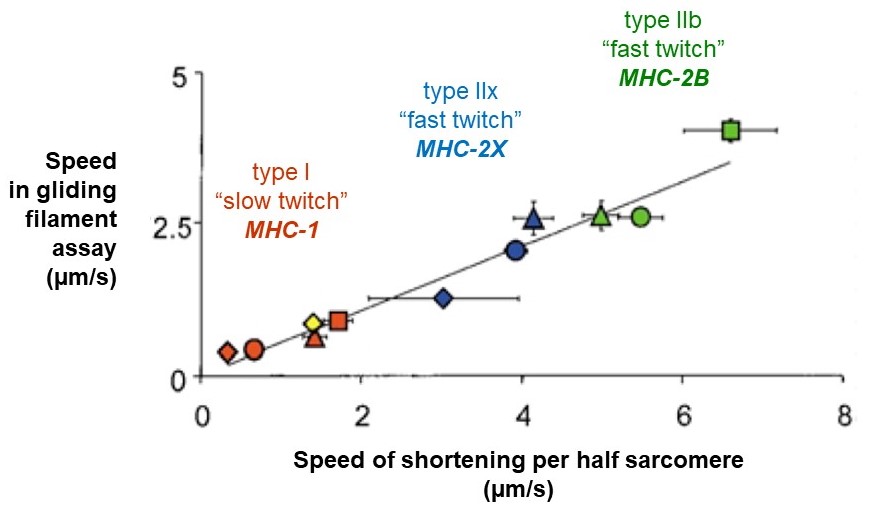
(MA Pellegrina 2003 J Physiol 546(3):677-89.)
Muscle fibers are often classified into more than two types. A common, three-type scheme designates three classifications, slow oxidative (Type I), fast oxidative (Type IIa), and fast glycolytic (Type IIb or IIx). These are based on twitch contraction speeds, on the primary isoform of myosin expressed in the fiber, and on the fiber’s capacity for oxidative phosphorylation (see metabolism sessions). In addition to variations in speed and myosin ATPase activity, the fibers can also differ considerably in their myoglobin content, glycogen stores, and rate of fatigue.
A note about exercise: Skeletal muscle responds to resistance training by becoming larger and stronger. Muscle enlargement and strengthening after exercise occurs by hypertrophy, an increase in the diameter of the muscle cells (fibers) – not by hyperplasia, an increase in the number of cells. Exercise can also cause minor changes in the myosin isoforms expressed within the fibers, causing minor changes in the fiber-type distribution of the muscle. For example, the fraction of fast oxidative type-IIa fibers might increase slightly, with a corresponding decrease in the fraction of fast glycolytic type-IIx fibers. However, these changes in myosin isoform expression are modest compared to the changes in cell diameter. Some smooth muscles do undergo hyperplasia, such as those in the uterus, which continue to grow in response to hormonal signals like estrogen, during puberty.
Simple exercises to check what you recall
The degree of control we have over the amount of force our skeletal muscles exert is exquisite. Using the very same muscles we can gently manipulate a delicate object or exert a crushing grip of 100 lbs or more. As you have learned, the neuro-muscular junction acts as a simple, binary relay – essentially every action potential that reaches the motor end plate elicits an action potential on the muscle fiber and therefore a twitch contraction. How then do we regulate muscle force? Muscle contraction can be adjusted in two ways: by changing the frequency of stimulation, and by recruitment of more and larger motor units.
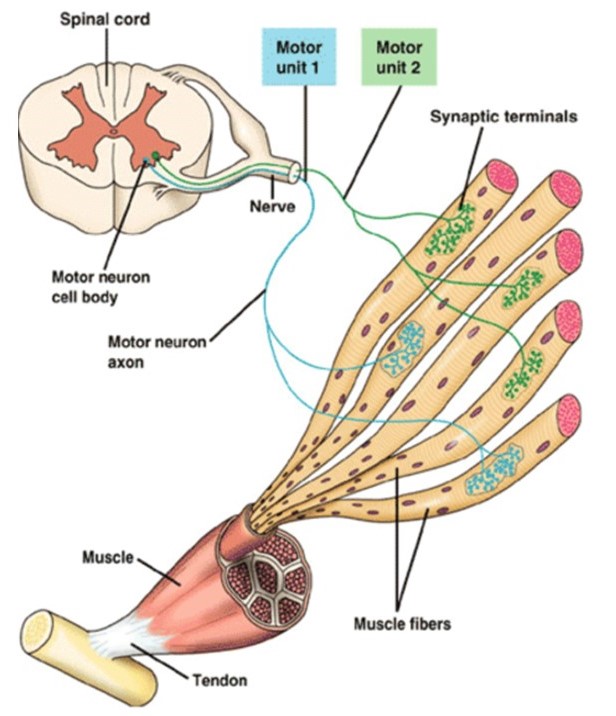
Motor units are quantal elements of muscle action. A skeletal muscle motor unit consists of a motor neuron and the several muscle fibers that it innervates (Figure 13). Upper motor neurons, from the primary motor cortex in the frontal lobe, synapse onto lower motor neurons in the brainstem or spinal cord to control voluntary movements. The details of these central nervous system pathways will be covered later, in your Mind Brain Behavior block. Here we focus on how motor units allow fine adjustments of muscle force.
Increasing the stimulus frequency can increase skeletal muscle force, but only modestly
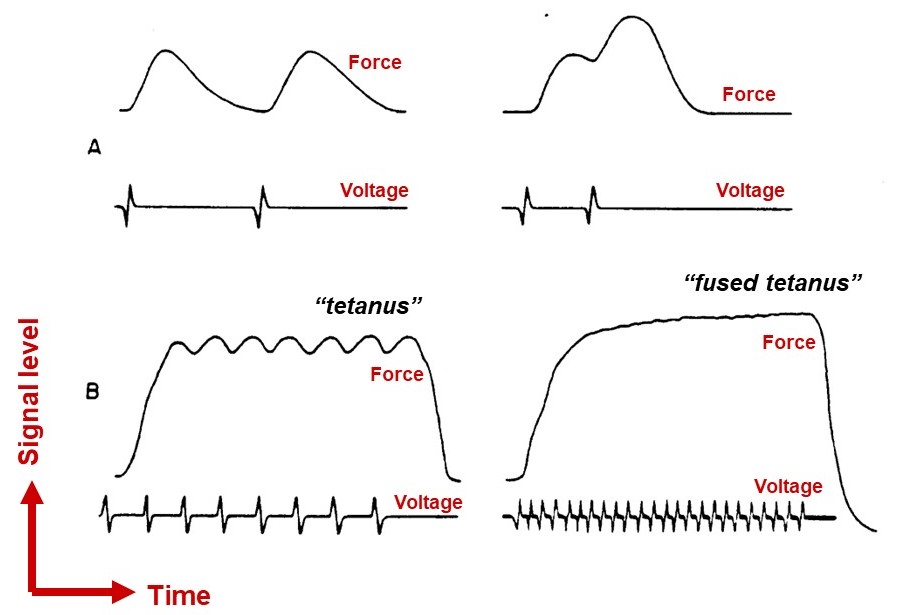
An action potential is all-or-none, but the force a muscle fiber develops as a result is not all-or-none. The force can be increased to some degree by increasing the frequency of action potentials, up until a fused tetanus is reached, where the fiber produces its maximal force (Figure 14). With faster stimulation, calcium release outpaces calcium sequestration, so more calcium builds up in the cytoplasm. Higher calcium allows a greater fraction of the myosins to bind F-actin, thereby allowing more force to be generated. Notably, however, this ‘temporal summation’ mechanism allows variation of muscle fiber force over only a limited range. Other mechanisms, which regulate the number of muscle fibers that are recruited during a muscle contraction, are required for fine control across a broad range of forces.
Regulation of skeletal muscle force occurs primarily by changes in motor unit recruitment
When the central nervous system sends stimulatory input to a particular muscle, the motor units innervating that muscle are recruited in an orderly manner, following the size principle: The first motor units to be activated are those with the smallest axons. These small units have relatively few, weak fibers and thus they generate the weakest contractile forces, allowing the total muscle force to be finely graded. As more units are recruited, motor neurons with progressively larger axons are activated, generating progressively larger amounts of force. This size-ordered recruitment enables fine motor control when little force is needed, and also enables very large forces to be generated when necessary.
Biophysics side note: A simple mechanism underlies size-ordered recruitment of skeletal muscle motor units: Motor neurons come in a range of sizes, some smaller and some larger in diameter. The smaller motor neurons have fewer parallel ion channels and thus higher electrical resistance (R). Synaptic input current (I) will thus generate bigger excitatory post-synaptic potentials in the smaller motor neurons (by Ohm’s Law, V = I·R), bringing them to threshold for action potentials sooner than larger motor neurons. The smaller motor neurons also happen to innervate fewer muscle fibers, so they elicit less force. Thus the neurons that are excited first are the ones that elicit the smallest (finest) increases in total muscle force.
Simple exercise to check what you remember
CARDIAC MUSCLE
Cardiac muscle cells (cardiac myocytes, or cardiomyocytes) are striated, just like skeletal muscle cells, because their contractile filaments are very similarly organized, into sarcomeres connected end-to-end to form myofibrils. Unlike skeletal muscle, however, cardiac muscle contracts involuntarily, in response to intrinsic pacemaker signals that are regulated by the autonomic nervous system. The molecular mechanisms underlying excitation-contraction coupling are very similar in cardiac and skeletal muscle, but there are a few key differences. To understand these differences, consider again the steps of excitation-contraction coupling, but focusing now on cardiac rather than skeletal muscle.
Voltage changes in the t-tubules of cardiomyocytes release calcium indirectly, via calcium-induced calcium release (CICR)
Like skeletal muscle, the t-tubules in a cardiac myocyte have voltage-sensitive channels, called Cav1.2, embedded in them. When an action potential arrives at a cardiac t-tubule, the voltage change likewise causes the Cav1.2 channels to open, allowing external calcium from the lumen of the t-tubule to flow into the cell. However, unlike in skeletal muscle, the Cav1.2 channels in cardiac myocytes are not directly linked to RyRs embedded in the SR. Cardiac and skeletal muscle cells contain different isoforms of RyRs. The cardiac RyRs are less abundant than in skeletal muscle, and they do not associate directly with Cav1.2 channels. Instead, external calcium entering a cardiac cell through Cav1.2 must bind to the RyRs to induce them to open – a process called calcium-induced calcium release (CICR). This indirect form of coupling has an important advantage over the more direct coupling found in skeletal muscle, because it allows graded regulation of the strength of heart muscle contractility by the autonomic nervous system. An action potential in a skeletal muscle cell nearly always releases a maximal amount of calcium from the SR, producing a twitch contraction of nearly uniform strength in every instance. By contrast, in a cardiac muscle cell, the amount of force is adjustable, via autonomic regulation of the amount of calcium released. Thus, calcium levels in cardiomyocytes serve not only as an “on-off switch”, but also as an adjustable “rheostat” that controls how forcibly the cell contracts.
Autonomic innervation regulates heart rate
The autonomic nervous system (ANS) regulates both the rate and forcefulness of cardiac muscle contraction. Heart rate is controlled by both parasympathetic and sympathetic innervation of the SA node, via modification of the rhythm of pacemaker cells in the node. The membrane potential of these cells oscillates in a regular, rhythmic pattern due to special properties of their ion channels, the details of which will be covered later, in your Circulatory Systems block. For now, understand that the parasympathetic vagus nerve innervates the pacemaker cells and acts to decrease their firing rate, while sympathetic nerves act to increase their firing rate. (Some details are summarized in the box below.)
Autonomic innervation also regulates the contractility of the heart
In addition to regulating heart rate, the ANS also controls the forcefulness (contractility) of cardiac muscle contraction. Cardiac contractility is regulated by the sympathetic nervous system, which innervates the entire heart. Sympathetic postganglionic fibers synapse onto the cardiomyocytes and release norepinephrine, which binds β1-adrenergic receptors to activate a signaling cascade (via Gαs). This G-protein-based cascade increases the amount of calcium released per heartbeat, and thus the contractility of the cardiomyocytes, in at least two ways (depicted in the interactive diagram below, and explained in detail in the following two paragraphs). Circulating epinephrine secreted by the adrenal medulla has the same effects.
β1-adrenergic stimulation increases contractility by at least two mechanisms, regulating the voltage-gated Cav1.2 channels that allow influx of external calcium, and also regulating the SERCA pumps that sequester calcium into the SR. Both mechanisms occur via activation of a Gαs pathway that you should recall from your earlier sessions on G-protein coupled receptors: Gαs activates the membrane enzyme adenylyl cyclase, which converts ATP into cytoplasmic cAMP (3’,5’-cyclic adenosine monophosphate), which in turn activates PKA (cAMP-dependent protein kinase). PKA directly phosphorylates Cav1.2 channels, thereby increasing their open-probability and allowing more external calcium to enter the cardiomyocytes during each heartbeat. Higher calcium results in more activation of thin filaments (see above, Step 5 of EC coupling in skeletal muscle), and thus a more forceful contraction.
PKA affects SERCA pumps too: Just as in skeletal muscle, the relaxation of cardiac muscle cells requires calcium to be returned to the SR by SERCA pumps. (Some calcium is also extruded to the outside of cardiac cells by sodium-calcium exchangers, but the SERCA pumps are responsible for removal of most of the calcium that enters during each contraction.) In cardiomyocytes, these SERCA pumps are regulated by another protein on the SR membrane called phospholamban. Phospholamban binds SERCAs and acts like a brake, partially inhibiting (slowing) their pumping action. The inhibition is relieved by PKA-dependent phosphorylation of phospholamban, thereby allowing SERCAs to pump more calcium into SR more quickly. When more calcium is pumped back into SR, more will be released from SR during the subsequent beat. Thus, the same sympathetic nerve input that increases heart rate by accelerating pacemaker activity in the SA node (as described in the previous section above), also increases the influx of extracellular calcium (via phosphorylation of Cav1.2 channels) and increases the speed and amount of calcium sequestration into SR (via phosphorylation of phospholamban). The overall result is a heartbeat that is both faster and more forceful.
SMOOTH MUSCLE
Smooth muscle surrounds hollow cavities and tubes in our bodies. At the most fundamental level, the contraction of smooth muscle is driven by essentially the same chemo-mechanical cycle of myosin as in skeletal and cardiac muscle (see Figure 5). However, smooth muscle is not striated like skeletal or cardiac muscle, because its contractile proteins are arranged less regularly. Smooth muscle drives constriction of blood vessels to control blood flow, contraction of the uterus during childbirth, contraction of the bladder (for micturation, a.k.a. peeing), peristaltic pumping (peristalsis) in the esophagus for swallowing and in the intestines during digestion and closing of various sphincters of the digestive and urinary tracts (e.g., pyloric sphincter at outlet of stomach, anal sphincters, internal urethral sphincter). Generally, these occur in response to involuntary signals from the autonomic nervous system. (The anus and urethra have both internal and external sphincters. Internal sphincters are subconscious and controlled by smooth muscle. External sphincters are conscious and controlled by skeletal muscle.)
Smooth muscle cells lack t-tubules and have less SR
Excitation-contraction coupling in smooth muscle differs substantially from skeletal or cardiac muscle. There are no t-tubules in smooth muscle cells, so action potentials can only propagate along their surface. They contain less SR and thus may not store enough calcium for full contraction. Instead, much of the required calcium enters from outside the cell, through voltage or ligand-gated channels in the plasma membrane. This external calcium must diffuse across the width of the cell to reach all the contractile machinery, limiting the speed at which full contraction can develop. Note how different this situation is from skeletal and cardiac muscle, where every sarcomere is surrounded by SR and can be quickly flooded with calcium that needs only to diffuse a very short distance. The slow response of smooth muscle cells to stimulation is not a disadvantage, since they generally drive processes that are slow.
Calcium controls smooth muscle myosin via a two-stage phospho-regulatory cascade
The contractile proteins in smooth muscle are activated by a very different, slower calcium-dependent mechanism than in skeletal and cardiac muscle. There is no troponin in smooth muscle, and thus the F-actin filaments are not regulated. Instead, it is the myosin that is regulated. (Note that different myosin isoforms are found in smooth, cardiac, and skeletal muscle.) Skeletal muscle myosin is always ready to cycle; but in resting smooth muscle, the myosin is in a quiescent state (unphosphorylated). Calcium entry into a smooth muscle cell initiates a cascade (depicted in the interactive diagram below) where calcium first binds calmodulin, and then calcium and calmodulin together bind a kinase enzyme, myosin light chain kinase (MLCK), activating it. Active MLCK phosphorylates myosin (on one of its two light chains) to initiate chemo-mechanical cycling of the myosin. Smooth muscle myosin is turned off by an enzyme, myosin phosphatase, that removes the activating phosphates. Notably, removal of phosphates from myosins that are already attached to F-actin slows (rather than accelerates) their detachment from the actin. This so-called latch (or latch-bridge) behavior is thought to help smooth muscles maintain luminal pressures (e.g., in the vasculature) more economically than they would otherwise, by allowing them sustain tension over long times while using less ATP.
A simple exercise to check what you recall
Can you identify similarities and differences in EC coupling across the three muscle types? Drag the features of EC coupling (gray boxes at bottom) onto the correct muscle types. Hints: Some features apply to more than one muscle type, and each muscle type should get four features (one from each column).
CARDIAC MUSCLE
Depolarization of cardiomyocytes occurs via gap junctions or pacemaker cells
Cardiac myocytes are linked end-to-end by intercalated discs, which allow mechanical forces to propagate from one cell to the next, and which also contain gap junctions (Figure 15). The gap junctions allow electrical currents and chemical signals to spread between neighboring cells, creating one functional unit composed of many interconnected cells. Note how this total interconnectedness is unlike the situation in skeletal muscle, where only individual cells (fibers) can be activated by a particular motor neuron. Cardiac muscle contraction is triggered by a set of pacemaker cells located on the right atrium of the heart, called the sinoatrial (SA) node, which generates action potentials rhythmically. (The basis for pacemaker activity in SA node cells will be discussed in more detail later, in your Circulatory Systems block.) When the command to contract is issued by the pacemaker, it spreads throughout the heart by the extensive branching of the connected myocytes.
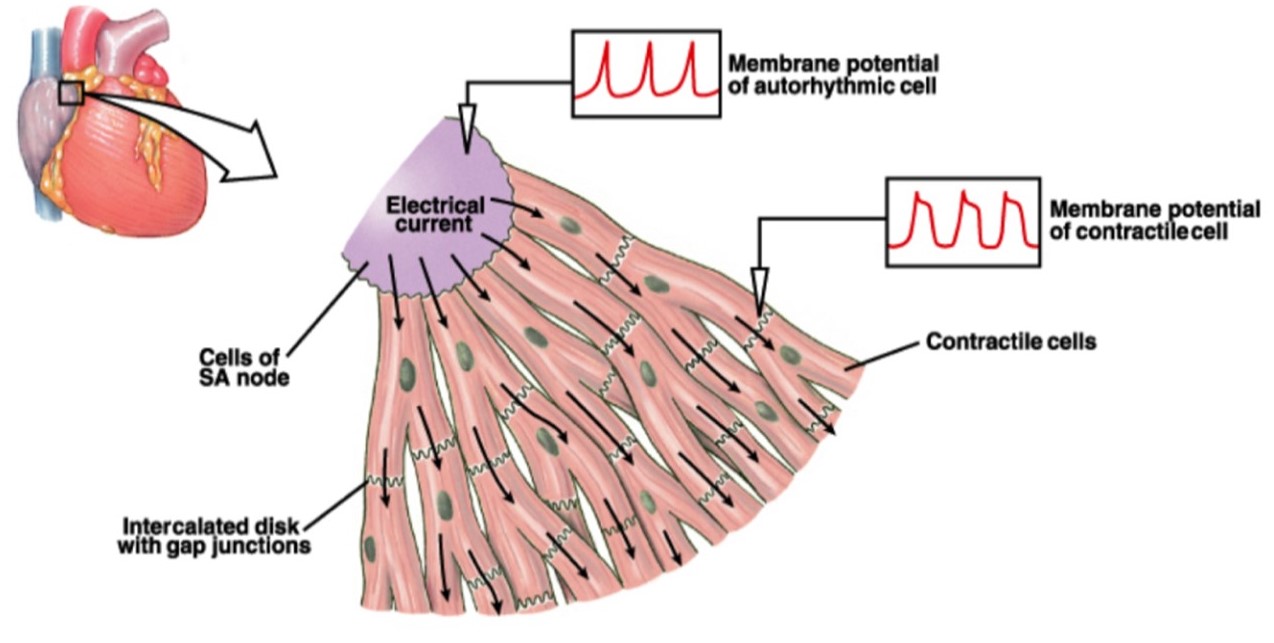
(From Silverthorn 2007 Human Physiology)
Just as in skeletal muscle, each cardiac muscle cell contains t-tubules continuous with the sarcolemma, although not as many, and an extensive network of internal sarcoplasmic reticulum (SR) calcium stores surrounding each sarcomere. Just as in skeletal muscle, the t-tubules in cardiac myocytes are located nearby the SR, allowing action potentials to penetrate the cell and deliver the “contract” signal nearly simultaneously to all the contractile machinery in the cell.
Cardiac muscle contraction always involves all fibers
The gradual recruitment of motor units in a skeletal muscle (described above under Learning objective #4) contrasts with what occurs in the heart. With each beat, all the cardiac muscle fibers in the heart are involved in generating force as a single unit. There is no option for recruiting additional motor units. Instead, to increase the forcefulness of the heartbeat, the calcium permeability of cardiac muscle fibers is regulated by the autonomic nervous system (as discussed above, under Learning objective #5).
In addition to their regulation by the autonomic nervous system, cardiac muscle fibers are intrinsically stretch-activated: Cardiac sarcomeres normally operate over a length range where the amount of active force they generate increases with their length. This stretch-activation of heart muscle was first discovered in 1914 and is known as the Frank-Starling Law or as Starling’s Law of the heart. It causes the heart to beat more forcefully when it becomes more filled, enabling it to automatically pump all the blood that returns to it, even in the absence of neuronal or hormonal input. (You will learn more about cardiac output and Starling’s Law later, in your Circulatory Systems block.)
SMOOTH MUSCLE
Different smooth muscles can be innervated in two distinct patterns, multi-unit or single-unit (Figure 16). Their innervation patterns influence how they behave.
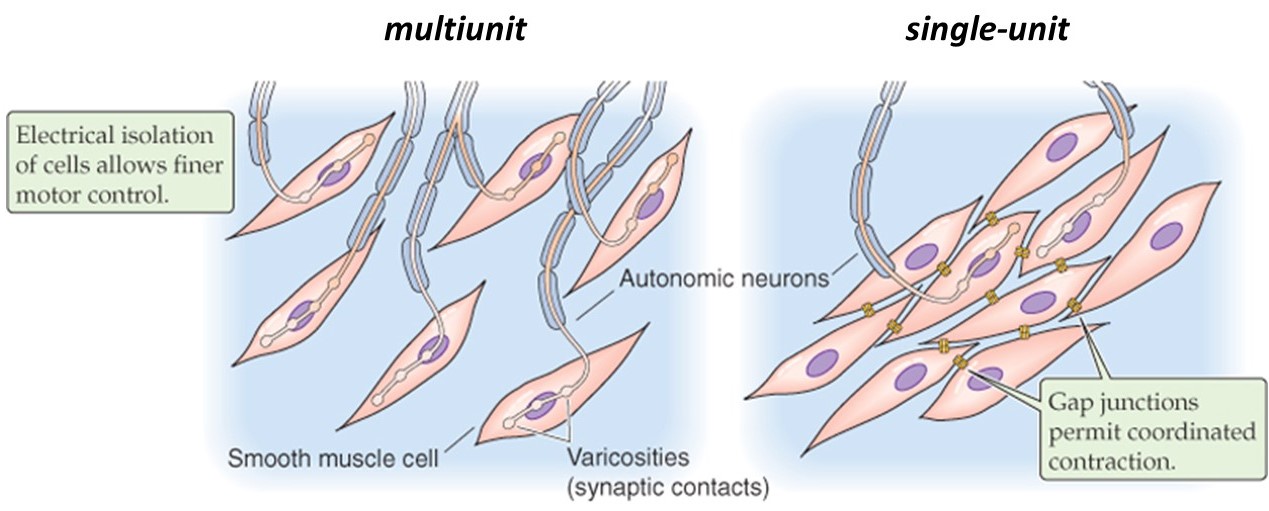
(Boron & Boulpaep 2009 Medical Physiology)
In a multi-unit smooth muscle, each cell is directly innervated by a post-ganglionic autonomic motor neuron, similar to the situation in skeletal muscle. Like in skeletal muscle, the innervation of individual cells in a multi-unit smooth muscle allows graded recruitment, and thus fine control of force generation. Because multi-unit smooth muscles are capable of maintaining sustained contractions, they are sometimes referred to as “tonic” smooth muscles. They are found for example in large airways, in the ciliary and iris muscles of the eye (which control focus and aperture, respectively), in sphincters, vasculature, and arrector pili (the tiny muscles that raise hairs and goose-bumps on your skin).
By contrast, the cells in a single-unit smooth muscle (also called unitary smooth muscle) are connected together by gap junctions, which allow electrical signals to spread from one cell to its neighbors. The interconnected cells act together as a single unit, with waves of electrical activity passing through them. Smooth muscles can contain pacemaker cells, similar to the SA node of the heart, and autonomic neurons may also synapse on a some of the cells to help regulate the contractility of the muscle. Because unitary smooth muscles contract transiently when stimulated, they are also known as “phasic” smooth muscles. They are found for example in the gastrointestinal tract and urogenital walls, where peristalsis is coordinated down the length of a tube.
Many smooth muscles are blends of both phasic and tonic, which allows them to respond to a range of stimuli. Smooth muscles in different body organs vary substantially in fiber organization, innervation and responsiveness to various stimuli.
A simple exercise to check what you recall
Drag each muscle type (gray boxes at right) to the category (1 or 2) where it belongs:
Extra Q&A from past students that might be helpful
Professor Asbury,
I was hoping to ask you one question in regard to the cardiac and smooth muscle. I understand that in skeletal muscle, we have a clear neuromuscular junction. However, for cardiac and smooth muscle, it seems it can be done via an electrical action potential (SA node pacemaker and nerve attachment respectively) and by G protein pathways (Gs and Gq respectively). Am I thinking of that the right way?
You’re absolutely correct about skeletal muscle, but you might be misunderstanding something about cardiac muscle: In cardiac muscle, contraction is *always* triggered by an electrical action potential. The action potential (AP) is delivered to cardiac cells in a different manner than delivery to skeletal muscle cells. For cardiac muscle cells, the AP is delivered by direct transmission from neighboring cells via gap junctions (and, ultimately, if you trace it back, the AP is initially created by the pacemaker cells in the SA node). But in both skeletal and cardiac, contraction is *always* triggered by an electrical AP.
What might be confusing is that cardiac muscle cells receive additional input from the autonomic nervous system (ANS). This ANS input does *not* trigger cardiac muscle contraction. Instead, it regulates how *forcefully* the cardiac muscle cells contract whenever they receive an AP from the pacemaker. The ANS input is received by GPCRs on the cardiac muscle cell surface (beta-1 adrenergic), then it gets relayed through G-alpha-s, and ultimately increases the forcefulness of the contractions elicited from each SA-generated AP (via increasing calcium influx through Cav1.2, and also by promoting faster SERCA pumping).
Smooth muscles are quite variable. In some cases, smooth muscle cells are directly electrically coupled to their neighbors via gap junctions, so APs propagate from cell-to-cell just like in heart muscle. These so-called “single-unit” smooth muscles can sometimes have pacemaker cells too, so that the whole muscle undergoes waves of contractions, again similar to the heart (for example, smooth muscles that drive peristalsis). In other cases, each individual smooth muscle is innervated by a single neuron. This arrangement is similar to skeletal muscle, except that the neurons will be ANS neurons, and the receptors will usually be GPCRs. And just to make smooth muscle even a little bit more complicated, contraction of some smooth muscles can be triggered by non-neural inputs. For example, stretch-activated channels in vascular smooth muscle cells can open to allow calcium in – this input is called “myogenic” because it’s generated intrinsically by the myocytes themselves. Another example is histamine, which can be released from immune cells and bind GPCRs on airway smooth muscles, promoting their contraction – this is a cause of asthma attacks.
Hi Dr. Asbury,
I’m confused about this except from the syllabus: “Phospholamban binds SERCAs and acts like a brake, partially inhibiting (slowing) their pumping action. The inhibition is relieved by PKA‐dependent phosphorylation of phospholamban, thereby allowing SERCAs to pump more calcium into SR more quickly. When more calcium is pumped back into SR, more will be released from SR during the subsequent beat.” Why would sequestering calcium increase contractility? Wouldn’t you want calcium to stick around so that more actin binding sites are exposed, and more are involved in contraction?
This is a very good question. It illustrates that you’re thinking carefully about the physiology of the heart. That last sentence in the passage quoted from the syllabus is important: “When more calcium is pumped back into SR, more will be released”. It is also helpful to understand that the effects of ANS on cardiac contractility happen over minutes, a time scale that is much longer than the duration of a single heartbeat. Two things happen simultaneously in response to PKA activation, (1) more opening of the Cav1.2 channels is promoted, and (2) SERCA activity is also increased. This means that over time, the heart cells are bringing in more calcium from outside, via number 1 above, and — during the relaxation phases of the heartbeat cycle — they are also stuffing the extra calcium into stores (into sarcoplasmic reticulum), via number 2 above. Due to this combination, and after some minutes of ANS input, the amount of calcium released from SR during the contraction phase of each beat will be greatly increased. The higher peak calcium levels in the sarcomeres will uncover more myosin binding sites on thin filaments, allowing more force to be generated.
Feedback:
muscle tissue that is anchored by tendons onto bone and directly innervated by motor neurons; used for voluntary, “on-command” control of locomotion, etc.
the muscle of the heart that contracts to pump blood through the body
muscle tissue surrounding hollow cavities and tubes in our bodies
a single muscle cell, very long and thin
special name for the plasma membrane of a muscle cell (sarco = Greek for "flesh"; lemma = "husk")
a series of sarcomeres linked end-to-end
elementary contractile unit of a myofibril
one of two types of filaments inside muscle sarcomeres; extend outward from the sarcomere center (from the M-line) toward the Z-discs; made primarily of myosin
one of two types of filaments inside muscle sarcomeres; extend inward from Z-discs toward the center of each sarcomere; made primarily of F-actin, together with regulatory proteins such as troponin and tropomyosin
one of three families of ATP-powered motor proteins; myosins move along F-actin; movement of myosin along F-actin drives shortening of all muscle types
physiological term for individual myosin motors inside muscle sarcomeres; myosin heads emanating from thick filaments "bridge across" to thin filaments
"rigor" is a physiological term for tight binding of myosin to F-actin in the absence of ATP; "rigor mortis" refers to postmortem muscle rigidity due to a lack of ATP. (Latin: rigor = "stiffness"; mortis = "of death")
a giant protein, greater than 1 µm in length, that functions as a molecular spring responsible for the passive elasticity of muscle.
plate-like structures that separate neighboring sarcomeres; Z-discs are also sometimes referred to as Z-lines
a fibrillar protein that binds thin filaments in muscle sarcomeres and, together with troponin, regulates their response to calcium
a protein complex located on thin filaments in skeletal and cardiac muscle, but not in smooth muscle; binds calcium and regulates contractile activity in response to calcium
the process by which electrical excitation of a muscle cell triggers its contraction; often abbreviated "EC coupling"
a chemical neurotransmitter in the central and peripheral nervous system; often abbreviated "ACh"
receptor proteins that respond to the neurotransmitter acetylcholine and also to the agonist nicotine
chemically induced change in voltage specifically at the motor end plate, which is the portion of the muscle-cell membrane at the neuromuscular junction; abbreviated EPP
electrical current flowing specifically through the motor end-plate (which is the portion of the muscle-cell membrane at the neuromuscular junction)
a network of membrane tubes connected to the sarcolemma that transmit action potentials deep into skeletal and cardiac muscle cells; "t" stands for "transverse" -- the tubules are perpendicular to the long-axis of the muscle fiber
special name given to the endoplasmic reticulum of a muscle cell; abbreviated SR
a voltage-gated calcium channel located in the t-tubule membranes of skeletal muscle cells
a calcium-release channel located in the sarcoplasmic reticulum; can be opened by the poison, ryanodine
a protein that uses energy from ATP hydrolysis to pump calcium into the sarcoplasmic reticulum (SR) of muscle cells (or into the endoplasmic reticulum of non-muscle cells)
enzyme that breaks down acetylcholine
physiological term describing how many muscle fibers are activated during a muscle contraction
physiological term for a single motor neuron plus all of the skeletal muscle fibers innervated by its axon terminals
physiological term meaning a sustained contraction when a muscle fiber is maximally stimulated by a very high rate of action potentials in its input motor neuron
the principle that motor units are recruited in a precise order according to the magnitude of their force output, with small units being recruited first; creates task-appropriate recruitment
a voltage-gated calcium channel located in the t-tubule membranes of cardiac muscle cells
cranial nerve responsible for parasympathetic control of the heart, lungs, and digestive tract
physiological term for the innate ability of muscle tissue to contract
a small protein that -- in its unphosphorylated form -- binds and inhibits SERCA
an intermediary protein that binds calcium and activates various calcium-sensitive enzymes
an enzyme that phosphorylates the light chain of smooth muscle myosin, thereby activating the motor
an enzyme that dephosphorylates the regulatory light chain of smooth muscle myosin, thereby de-activating the motor
a small group of specialized cells in the right atrium of the heart that are responsible for initiating the heartbeat
a type of smooth muscle in which the cells are not connected by gap junctions. As a result, electrical excitation does not spread from one cell to the next, instead remaining confined to the cell that was originally stimulated
a type of smooth muscle in which neighboring cells are connected by gap junctions to synchronize their membrane depolarization and contractions; so the muscle contracts as a single unit
