
Receptor tyrosine kinases and nuclear hormone receptors
Mark Bothwell
Introduction
After G protein-coupled receptors, receptor tyrosine kinases and nuclear hormone receptors are the two largest families of receptors mediating cell-cell communication. Receptor tyrosine kinases (RTKs) are receptors for a variety of polypeptide hormones and growth factors. Nuclear hormone receptors are receptors for steroids and other non-peptide hormones. RTKs and nuclear hormone receptors often mediate effects that are slower in onset but more lasting in duration than GPCRs, but like GPCRs they typically initiate cascades of events that change the physiological state and patterns of gene expression of cells.
Session Learning Objectives
1. Describe the structure and function of RTKs.
2. Describe signaling mechanisms used by RTK signaling (MAPK cascades).
3. Describe crosstalk and interactions between pathways.
4. Describe steroid hormone receptors.
5. Describe steroid hormone receptor mechanisms of activity.
RTK Structure and Function
Receptor tyrosine kinases (RTKs) signal across the plasma membrane in response to binding of growth factors, hormones such as insulin, and other secreted polypeptides. RTK proteins are large intrinsic membrane proteins possessing a single membrane-spanning segment. The receptors function as dimers of two identical subunits.
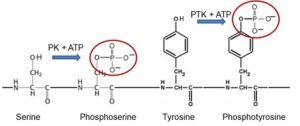
Protein kinases represent one of the most important classes of proteins engaged in cellular regulation. Over 500 human genes encode protein kinases. While the majority of protein kinases phosphorylate the hydroxyl groups of protein serine or threonine residues (Fig. 1), about 90 human genes encode protein kinases known as tyrosine kinases because they phosphorylate the hydroxyl groups of protein tyrosine residues. More than half of tyrosine kinases are referred to as receptor tyrosine kinases (RTKs) because their tyrosine kinase enzymatic activity is activated by binding of extracellular peptide hormones such as insulin or of growth factors such as nerve growth factor (NGF), epidermal growth factor (EGF) and vascular endothelial growth factor (VEGF).
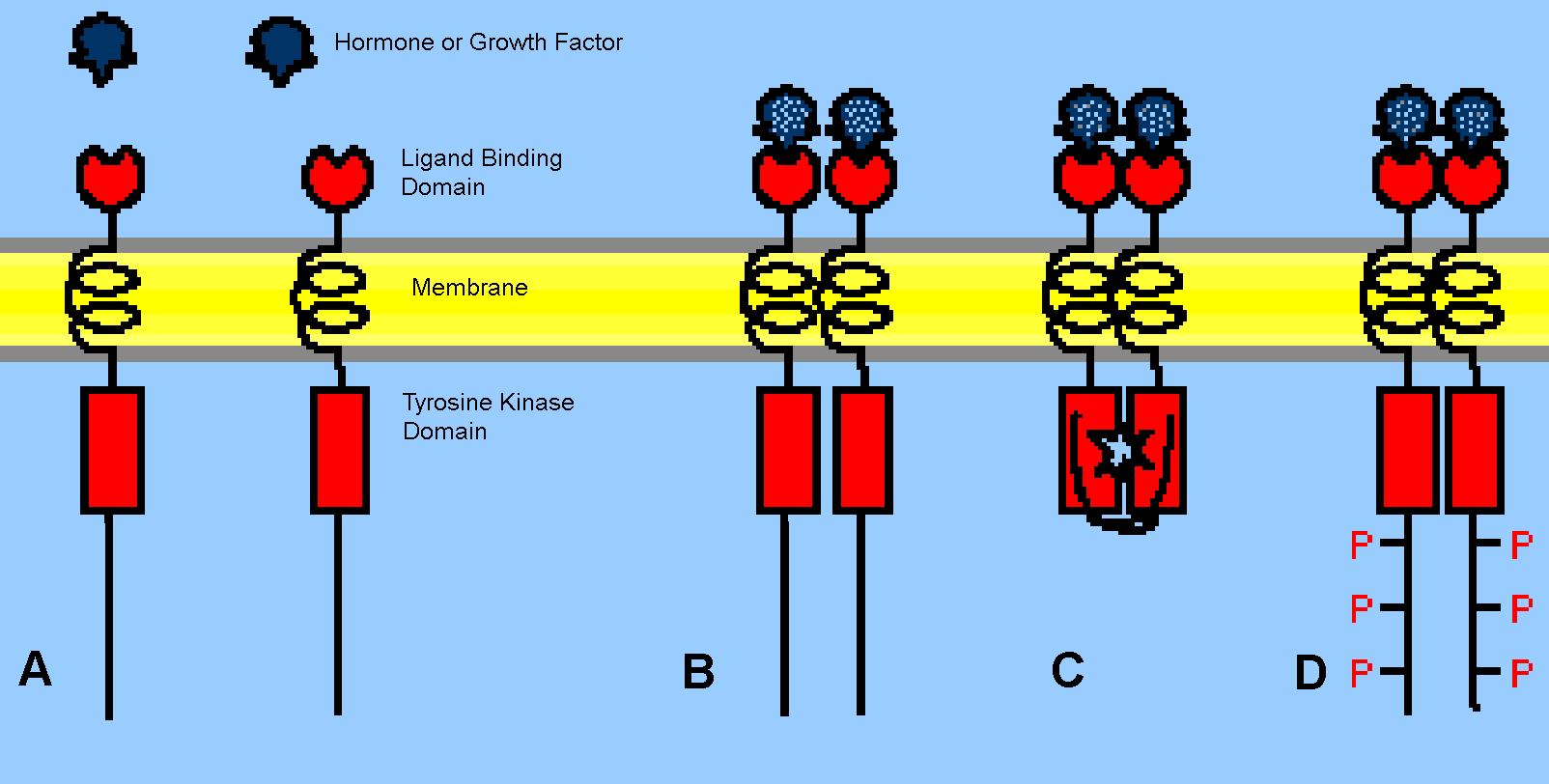
RTKs span the plasma membrane, so that hormone or growth factor binding to the portion of the receptor exposed to the extracellular environment stimulates the tyrosine kinase activity of the portion of the receptor exposed to the cytoplasm (Fig. 2A).
Some RTKs exist constitutively as dimeric aggregates, while others form such dimers in response to ligand binding (Fig. 2B). In the ligand-activated dimer, each subunit phosphorylates tyrosine residues in its partner subunit, a process known as auto-phosphorylation. In contrast to the serine-threonine kinase activity of PKA or PKC, the tyrosine kinase activity of RTKs is directed against a relatively smaller number of substrates, most notably the protein chains of the receptor itself.
A simple exercise to check what you recall
[h5p id=”36″]
Most signaling from RTKs occurs by nucleation of protein complexes scaffolded on the phosphorylated tyrosine residues (phosphotyrosines) of activated RTKs (Fig. 1 and 2D). The phosphotyrosines are recognized as docking sites by different cytoplasmic signaling proteins, initiating several parallel signaling pathways.
Ras/ERK Pathway
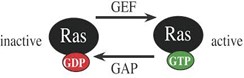
One important signaling pathway initiated by RTK auto-phosphorylation employs the monomeric G protein, Ras. Like heterotrimeric G proteins, monomeric G proteins cycle between an activated state, with GTP bound, and an inactive state, with GDP bound (Fig. 3), and the activated form binds effector proteins to cause downstream events. The activity of monomeric G proteins is controlled by (i) GTP Exchange Proteins (GEFs) that promote exchange of bound GDP with GTP, increasing Ras signaling activity, and (ii) GTPase Activating Proteins (GAPs) that promote the intrinsic GTPase activity of Ras, converting bound GTP to GDP and turning off Ras signaling activity. Unfortunately, a variety of human Ras gene mutations dysregulate Ras. Elevated cell proliferation associated with activating mutations of Ras drives one third of human cancers.
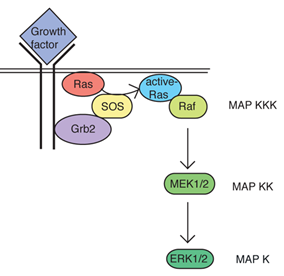
A key GEF controlling Ras activity is the protein called SOS. SOS is turned on by binding to an adaptor protein Grb2 that associates with particular phospho-tyrosine residues of activated RTKs (see Fig. 4). Thus, activation of RTKs can promote activation of Ras through Grb2 and SOS acting as adaptors. This is an example of formation of a larger intracellular signaling complex.
A common functional theme among monomeric G proteins is cascading activation of a series of three serine/threonine-directed protein kinases (Fig. 4). The final protein kinase in the series is a Mitogen Activated Protein Kinase (MAPK). MAPK becomes active as the result of phosphorylation by a MAPK kinase (MAPKK) and the MAPKK becomes active following phosphorylation by a MAPKK kinase (MAPKKK). MAPKKK is activated by binding to a monomeric G protein such as Ras.
In the case of Ras, the MAPKKK is a protein named Raf, the MAPKK is MEK1 or MEK2, and the MAPK is ERK1 or ERK2. ERK1 and ERK2 typically function redundantly so they are often referred to jointly as ERK1/2. ERK1/2 signals in both the cytoplasm and nucleus. Among other important functions, ERK1/2 is a master regulator of the cell cycle. Thus ERK1/2 activity resulting from growth factor activation of RTKs promotes cell proliferation and tissue growth and repair. Constitutive activation of ERK1/2 is the primary driver of cell proliferation in most cancers. ERK1/2 activity also reduces cell death. Reduced cell death enhances the ability of increased cell proliferation to increase tissue mass, contributing to the tumorigenic effect of constitutively activated ERK1/2. Constitutive activation of ERK1/2 in cancer usually results from activating mutations of RTKs and/or Ras rather than mutations of ERK1/2 themselves.
Two simple exercises to check what you recall
[h5p id=”35″]
[h5p id=”34″]
PI3K/Akt Pathway of RTK Signaling
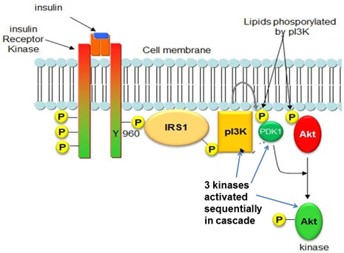
Some phosphotyrosine residues of RTKs bind and activate a phospholipid-modifying enzyme, phosphatidylinositol 3-kinase (PI3K), triggering another cascading sequence of kinases. For example, insulin receptor signaling employs a variant version of this mode of signaling (Fig 5). A phosphotyrosine residue of the activated insulin receptor binds a protein called IRS1. The insulin receptor then phosphorylates tyrosine residues of IRS1, causing IRS1 to bind and activate PI3K. The PI3K enzyme adds an additional phosphate group to phosphatidylinositol-4,5-bis-phosphate (PIP2), producing phosphatidylinositol-3,4,5-trisphosphate (PIP3). A protein kinase, Akt, is recruited to the plasma membrane by binding to PIP3, allowing Akt to be phosphorylated and activated by several different membrane-associated protein kinases. Activated Akt controls both cytoplasmic and nuclear functions. For example, insulin receptor-mediated activation of Akt acts in the cytoplasm to cause plasma membrane insertion of the glucose transporter, Glut4, allowing cellular uptake of glucose, while Akt acts in the nucleus to activate the transcription factor Fox01, which controls enzymes for hepatic glucose synthesis.
Phospholipase C (PLC) Pathway
Phosphotyrosine residues of some RTKs bind and activate [the gamma isoform of] Phospholipase C (PLC). The PLC enzyme activated by RTKs and the PLC activated by GPCR signaling are products of different genes, but they catalyze the same reaction, cleaving the lipid PIP2 to produce IP3 and DAG second messengers, increasing cytoplasmic calcium ion concentration, and activating protein kinase C (PKC), respectively. This pathway represents an important mode of convergence of RTK and GPCR signaling pathways.
Convergence and Cross-talk between RTK and GPCR pathways
An important feature of regulatory biology is that distinct pathways controlling similar functions may be linked in a manner that coordinates their functions. Convergence, as seen for PLC in GPCR and RTK pathways, represents one mode of linkage of RTK and GPCR signaling. Cross-talk represents another. The concept of cross-talk is illustrated by two mechanistically different examples.
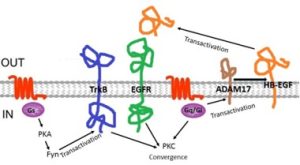
Example 1: The epidermal growth factor (EGF) receptor is an RTK that is activated by a variety of different growth factors (Fig. 6, green). Some EGF receptor ligands are released from cells by vesicular secretion, but others, such as HB-EGF (orange), are initially expressed as intrinsic membrane proteins that must be proteolytically cleaved by ADAM17 to release the active extracellular ligand peptide. Both Gi and Gq GPCR pathways activate ADAM17. Thus, GPCR ligands often cause EGF receptor activation. This type of crosstalk is known as transactivation.
Example 2: A second type of crosstalk/transactivation mechanism is illustrated by the Trk family of RTKs (Fig. 6, blue) that function as receptors for the nerve growth factor family of growth factors [known as neurotrophins]. GPCRs that activate the cAMP/PKA signaling pathway cause phosphorylation and activation of Fyn, a protein kinase. Activated Fyn binds and induces signaling by the TrkB neurotrophin receptor, even in the absence of neurotrophins. Thus, GPCR ligands such as adenosine can cause transactivation of TrkB signaling in neurons.
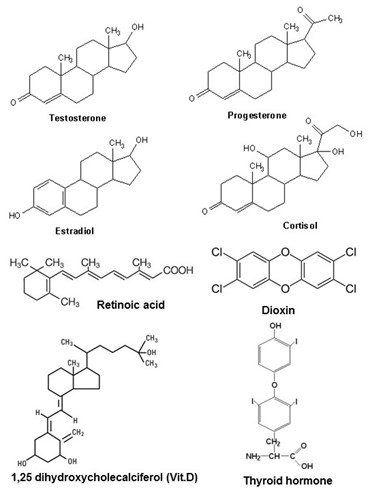
Steroid receptors are a subfamily of a larger group of receptors known as nuclear hormone receptors. The ligands for nuclear hormone receptors are structurally and functionally diverse, as illustrated in Figure 7, including hormones such as steroid hormones, thyroid hormone, and the hormonal derivatives of vitamin D and vitamin A, but also including aromatic hydrocarbons including the potent environmental toxin, dioxin. By exerting hormone-like effects, environmental dioxin pollutants cause reproductive and developmental problems, and cancer.
Ligands for nuclear hormone receptors all have one important property in common; they are chemically hydrophobic. This allows them to pass easily across cellular membranes. Their receptors reside in the interior of cells, shuttling between the cytoplasm and nucleus. The various steroid hormone receptors and the other nuclear hormone receptors are closely similar in structure, and they all signal in much the same way.
A simple exercise to check what you recall
[h5p id=”33″]
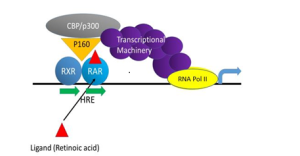
Unlike GPCRs and RTKs, nuclear hormone receptors are not associated with membranes. Steroid receptors and other nuclear hormone receptors signal by acting as transcription factors that regulate expression of specific genes (Fig. 8). They bind directly to short (6-nucleotide) DNA sequence motifs in the promoters of steroid-regulated genes. These sequence motifs are known as hormone response elements, abbreviated as HRE in the figure. [More specifically, scientists distinguish retinoic acid response elements (RRE), steroid response elements (SRE) and glucocorticoid steroid response elements (GRE), for example].
Binding of steroids to their receptors increases the affinity of the receptors for the cognate DNA sequence motifs in the gene promoter. The promoter-receptor-steroid complex promotes expression of the regulated gene. Following binding of the hormone/nuclear hormone receptor complex to DNA, the receptor recruits other transcriptional regulatory proteins, including RNA Polymerase II, allowing transcription of the gene to commence (Fig. 8). However, in some cases steroid receptors bind the promoter in the absence of steroid. In such cases the bound receptor actively represses gene expression, until binding of steroid to the receptor switches it from a repressive to an activating mode of action.
Nuclear hormone receptors function as dimers. Their dimeric structure allows them to bind optimally to sequence elements in both strands of the DNA double helix. Steroid receptors generally function as homodimers (two identical subunits in the complex). On the other hand, some nuclear hormone receptors function as heterodimers (two non-identical subunits). Figure 8 illustrates such a scenario for the retinoic acid receptor (RAR), which functions as a heterodimer with a second subunit known as retinoic X receptor (RXR). RXR also acts as the heterodimeric partner of other nuclear hormone receptors, including thyroid hormone receptor and vitamin D receptor.
Conclusion
An important feature of signaling by steroid receptors and other nuclear hormone receptors is that binding sites on DNA for each receptor appear in dozens or even hundreds of different genes. Thus, hormones such as estrogen and thyroid hormone coordinately change expression of many different genes and change many aspects of cell function in many different tissues.
