
4 Cystic Fibrosis
Introduction
Cystic fibrosis is an autosomal recessive genetic disease[1] that is due to mutations that reduce or eliminate function of the protein CFTR. As you have learned, the movement of Cl– ions through the CFTR channel (located on the apical membrane) is the rate-limiting and regulated step for fluid secretion by many epithelia. When CFTR is defective or absent in cystic fibrosis, it causes defective epithelial fluid secretion, causing pathology and tissue destruction, particularly in the digestive tract and lungs.
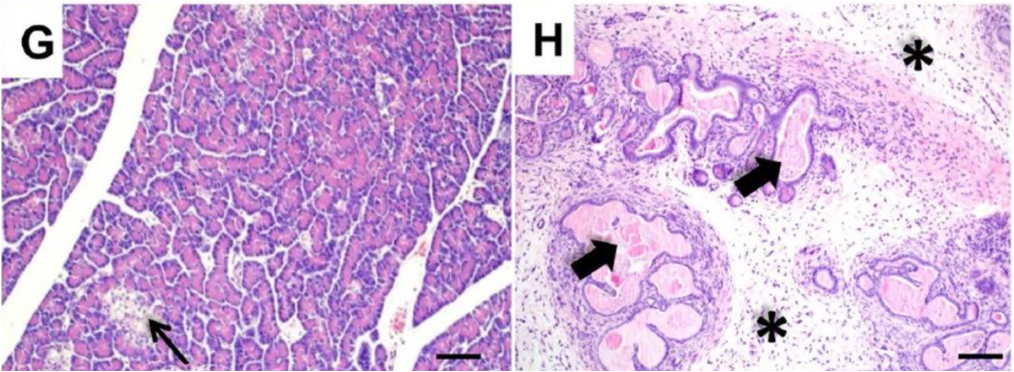
Cystic fibrosis was given its name by Dorothy Hansine Andersen, who first described the disorder in 1938. She performed microscopic examination of postmortem tissues from children who died from malnutrition in early infancy due to an unknown defect in digestion and absorption. The term “cystic fibrosis” describes the appearance of the pancreatic tissue from these patients: there were cysts (fluid filled spaces) due to dilated ducts, and fibrosis, which resulted from the replacement of normal pancreatic tissue with scar tissue. She found that in cases where she saw cystic fibrosis of the pancreas, it was associated with pathological changes in the lungs.
https://www.ncbi.nlm.nih.gov/pmc/articles/pmid/26365583/
Cystic Fibrosis Pathology in the Digestive Tract
Although most people think of cystic fibrosis primarily as a lung disorder, it affects secretion in the GI tract and thus causes digestive system pathologies. In the intestines, the lack of fluid secretion can cause intestinal blockage. For instance, some infants with cystic fibrosis present with meconium ileus, which is the failure to pass meconium (meconium refers to the first tarry stool produced by newborns).
The pancreas is an important location for CFTR-dependent secretion. Pancreatic secretion can be divided into endocrine secretion, in which hormones are secreted to the bloodstream, and exocrine secretion, in which fluid, bicarbonate, and digestive enzyme precursors are released into the small intestine.
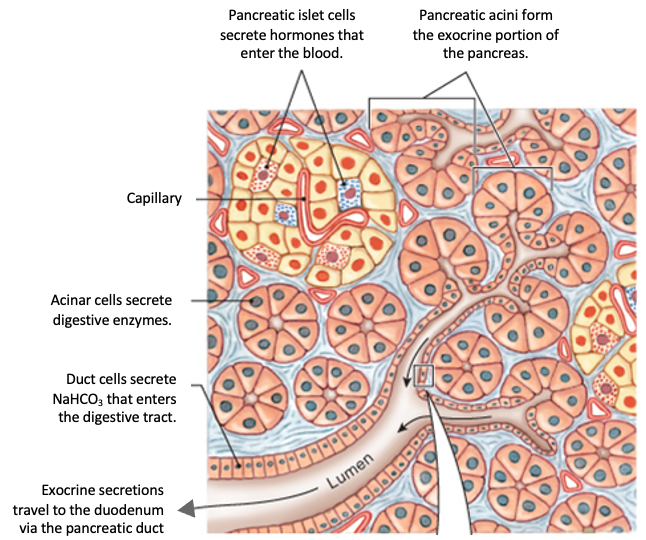
Clusters of cells called acinar cells secrete the inactive digestive enzyme precursors, which are known as zymogens. Duct cells are epithelial cells that form the small pancreatic ducts and the lining of larger ducts. Duct cells secrete bicarbonate ions and fluid.
Secretion by the duct cells is necessary to ensure that the zymogens are carried out of the pancreas and into the small intestine before being activated. In patients with a defective CFTR protein, the lack of secretion by pancreatic duct cells can block the ducts and lead to chronic pancreatitis, when inappropriate zymogen activation inside the pancreas leads to digestion of cellular components and subsequent inflammation, damage, and loss of pancreatic tissue. Chronic pancreatitis can affect individuals that are heterozygous for a CFTR mutation. (Heterozygotes have one normal CFTR allele, but have reduced CFTR function).
The most common digestive system pathology affecting patients with cystic fibrosis is pancreatic insufficiency, meaning there is inadequate production of digestive enzymes. This occurs because blocked ducts and tissue damage in the pancreas will prevent release of adequate amounts of digestive enzymes into the small intestine. If severe enough, pancreatic insufficiency causes malnourishment–for instance in an infant, it might cause a failure to thrive. Roughly 85% of newborns with cystic fibrosis have pancreatic insufficiency at birth or will develop it within the first year of life. Pancreatic insufficiency is treated with pancreatic enzyme replacement therapy. Below is a flow chart that details the steps involved in the development of pancreatitis and pancreatic insufficiency in a patient with cystic fibrosis.
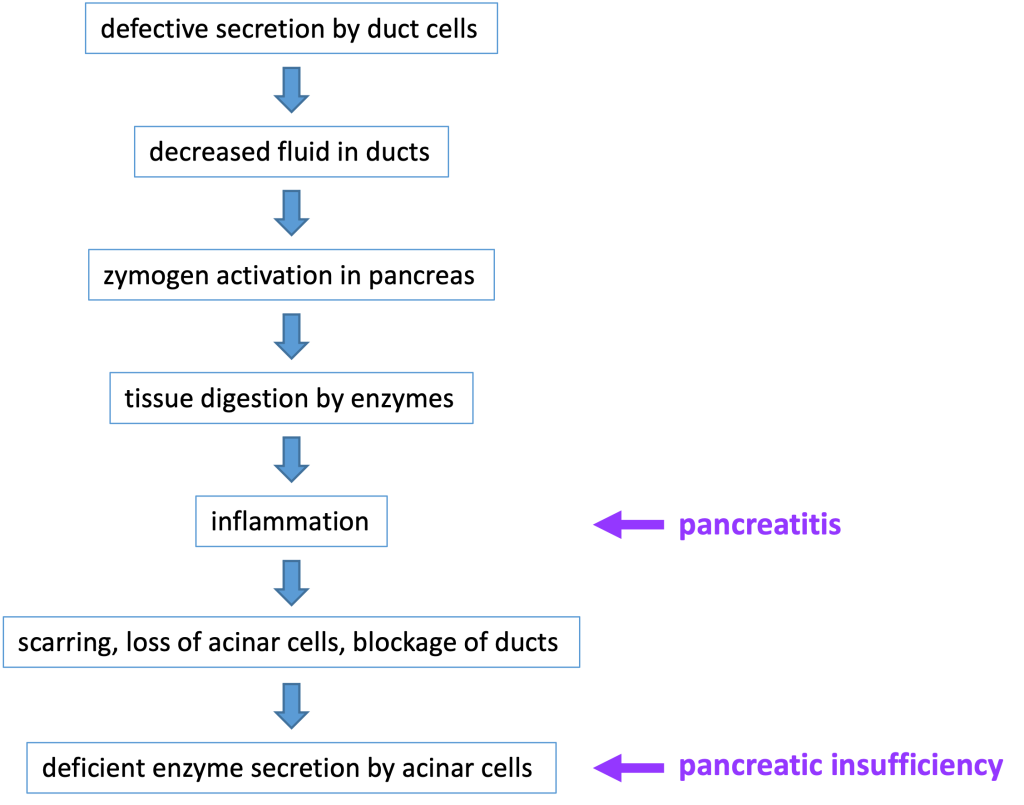
Because chronic pancreatitis causes generalized tissue damage in the pancreas, there may also be damage to endocrine tissue in the pancreas. When cystic fibrosis-associated pancreatitis damages the beta cells in the pancreatic islets of Langerhans, this causes insulin deficiency and diabetes mellitus. CFTR-related diabetes mellitus is treated with insulin replacement.
Cystic Fibrosis Pathology in the Lungs
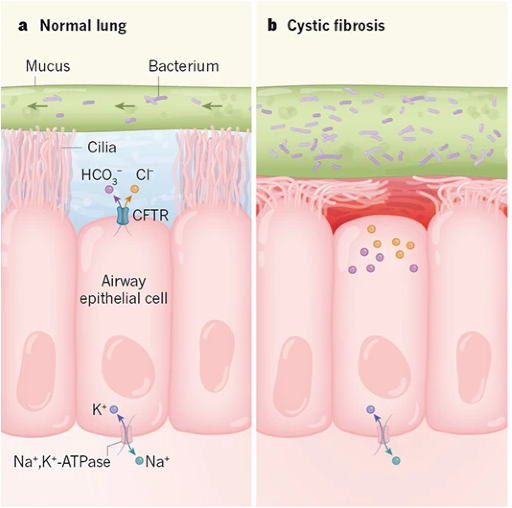
In the lungs, the defect in CFTR changes the environment in the airways so that they are much more prone to infection by pathogens. One important factor is that a lack of fluid secretion causes insufficient hydration of mucus. As a result, the mucus becomes very thick.
https://www.nature.com/articles/d41586-019-00781-y
Mucus is moved up and out of the lungs by the beating of cilia on the apical the surface of airway epithelial cells: this is called mucociliary clearance. Normally, the cilia are surrounded by a watery saline layer due to secretion by airway epithelial cells. In cystic fibrosis, because of defective secretion by airway epithelial cells, there is no watery saline layer and the thick mucus is not easily cleared. The thick mucus in the lungs of a CF patient is conducive to bacterial and fungal growth. This causes cycles of infection, inflammation, and tissue damage in the upper airways, which leads to further problems in clearing the mucus. However, the situation in the lungs is complex, and other changes caused by CFTR dysfunction may be contributing to pathology in the lungs.
Diagnosis and Treatment
More than 2000 different genetic mutations in the gene for CFTR have been identified. Biochemical and DNA testing is used to screen newborns to identify patients so that appropriate treatment can begin at an early age. One of the tests used to diagnose cystic fibrosis is the sweat chloride test[2].
The lung problems in cystic fibrosis have traditionally been treated with antibiotics to prevent infection and various therapies to hydrate and remove the thick mucus. An important innovation has been the development of inhaled antibiotics. Pancreatic insuffiency is treated with pancreatic enzyme replacement therapy.
The CFTR gene was identified in 1989. When early trials of gene therapy were unsuccessful, a program was launched to identify small molecule drugs that could modulate the function of defective CFTR proteins (CFTR modulators). Two general types of drugs have been developed.
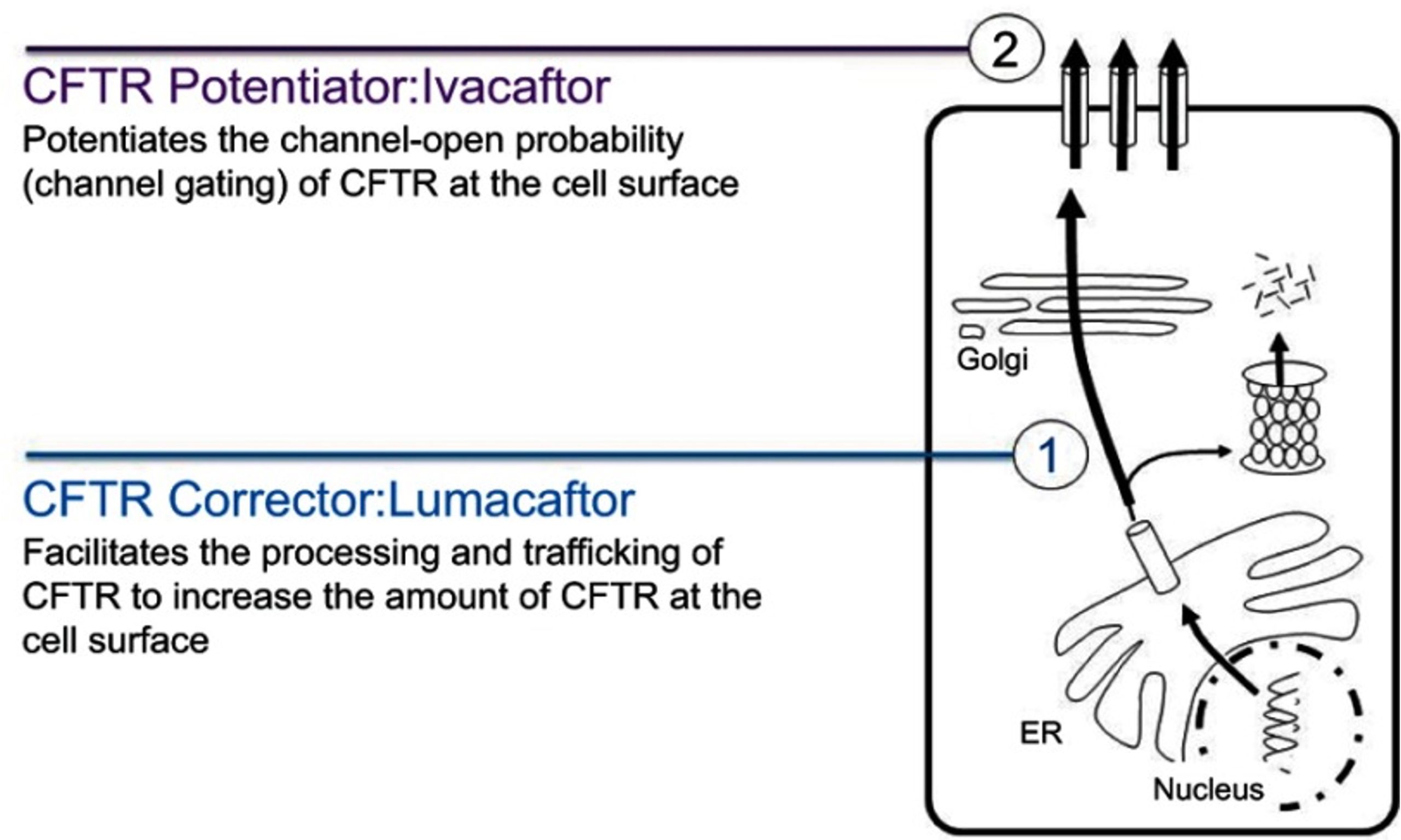
https://www.ncbi.nlm.nih.gov/pmc/articles/pmid/31409974/
Many CFTR mutations (such as the F508 deletion that affects two-thirds of all patients) affect protein trafficking of the mutant CFTR so that it doesn’t get expressed on the apical plasma membrane. CFTR correctors are drugs designed to improve protein folding and correct for this protein trafficking defect.
CFTR potentiators are drugs that improve CFTR function by increasing the amount of time that a mutant channel stays open.
The newest cystic fibrosis treatment, Trikafta (elexacaftor-tezacaftor-ivacaftor; approved in October 2019) is a combination of two CFTR correctors and one CFTR potentiator. Treatment with Trikafta should be able to effectively improve lung function in about 90% of patients with cystic fibrosis.
- In an autosomal recessive disease, the disease is expressed when both copies of the gene are mutant. ↵
- Interestingly, the epithelial cells in the sweat ducts are involved in the reabsorption of Cl- ions. The fluid secreted by the sweat glands is isotonic with the extracellular fluid, but as it flows through the sweat ducts, Na+ and Cl- ions are reabsorbed, so that the resulting sweat has a lower salt content. When CFTR is defective in cystic fibrosis, the chloride content of sweat is abnormally high. ↵