
4 Juvenile Idiopathic Arthritis (JIA)
- Overview:
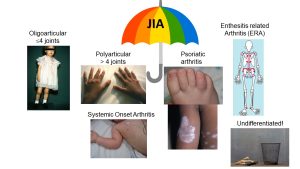
Juvenile Idiopathic Arthritis (formerly called juvenile rheumatoid arthritis or JRA) is an umbrella term for a number of heterogeneous categories of chronic immune-mediated arthritis seen in children. It is estimated that 1/1,000 children under the age of 16 have a form of JIA, making this disease as common as type 1 diabetes mellitus and more common than cystic fibrosis in childhood. There are 6 subtypes of JIA based on current clinical classification criteria (Oligoarticular-JIA, Polyarticular-JIA, Enthesitis-related arthritis, Psoriatic arthritis, Systemic onset (Still’s Disease) Arthritis and Undifferentiated JIA). Certain JIA categories are similar to corresponding adult-onset diseases and represent an early age of onset of the adult disease variant (ie. rheumatoid factor positive poly-JIA is thought to be akin to adult RA; Enthesitis-related arthritis = juvenile onset spondyloarthropathy; psoriatic arthritis in childhood is similar to psoriatic arthritis in adulthood; Systemic onset arthritis presenting after age 16 is known as Adult Onset Still’s disease) but several seem to be unique to childhood (oligo-JIA, rheumatoid factor negative poly-JIA).
- Clinical Presentation:
While some of the clinical and pathological disease features seen in JIA patients are similar to those seen in adult RA, it is important to remember certain features that are unique to arthritis in childhood. Patients often under report pain and often presenting symptoms are altered gait, limping, am stiffness and avoidance of usual activities. Systemic onset JIA (SJIA) is unique in presenting with daily high spiking fevers, rash, lymphadenopathy, and hepatosplenomegaly; arthritis in SJIA may be minimal and or present after the onset of systemic features.
- Pathophysiology:
Immune dysregulation leads to synovitis and without treatment will progress to eventual joint destruction. In systemic JIA, cytokines like IL-1 and IL-6 play a significant role. SJIA is unique from other forms of JIA in the tendency to be complicated by Macrophage activation syndrome (MAS) a life-threatening form of cytokine storm or hyperinflammation. MAS is type of secondary hemophagocytic lymphohistiocytosis (HLH) and is characterized by persistent fevers, cytopenias, hyperferritinemia, splenomegaly and other laboratory abnormalities. MAS often presents as a critical illness and requires prompt initiation of aggressive immunosuppressive therapy to avoid multi-organ dysfunction and death.
- Diagnosis:
Diagnosis is based on clinical presentation. All forms require:- Onset of symptoms prior to age 16
- Objective findings of arthritis lasting > 6 weeks (except SJIA)
- Exclusion of other causes
Additional features such as number and distribution of joints, and family history determine the subtype (Those details are beyond what is needed for MJBS)
Laboratory markers of adult RA disease, including anti-CCP and rheumatoid factor are frequently negative in JIA. These labs are sent as prognostic indicators; when positive they may indicate likelihood of response to certain medications and patterns of joint involvement. While certain JIA categories tend to present with an elevated ESR and CRP (in particular SJIA), these tests may also be normal to only mildly elevated in other patients. A mild anemia of chronic disease may be the only finding in certain patients. Radiographs are often normal unless there has been longstanding untreated disease. Ultrasound and MRI are useful for detecting synovitis.
- Treatment:
NSAIDs, DMARDs (methotrexate), biologics (IL-6 inhibitors, TNF-alpha inhibitors, IL-1 inhibitors), corticosteroids for severe cases, and physical therapy and routine ophthalmologic evaluations with a slit-lamp exam.
- Complications:
Similar to other forms of inflammatory arthritis, long standing inflammation can lead to bony erosions and joint destruction. However, there are also unique consequences of untreated arthritis in childhood; involvement of the growing skeleton may lead to impaired linear growth or growth deformities such as leg length discrepancy (for example, one leg grows longer than the other due to inflammation of one knee), and micro/retrognathia (undergrowth of the jaw due to involvement of the temporal-mandibular joints TMJs). Young, JIA patients (< 6 years old) with the oligo-articular form in particular are at high risk for a chronic asymptomatic form of uveitis (inflammation of the anterior chamber of the eye). Testing for ANA positivity can help to stratify risk – with ANA positive patients having a higher risk of eye involvement and needing more frequent ophthalmologic monitoring. If untreated, JIA-associated uveitis can lead to glaucoma, cataracts or permanent vision loss. This is one of the most common causes of acquired blindness in the US.