
11 Heme Therapeutics
Student Learning Objectives
- Explain the mechanism of action of therapeutics used clinically for non-malignant blood disorders that include anemia, sickle cell disease, ITP, and PNH.
- Recognize the clinical uses of hematology therapeutics in non-malignant hematology.
- Describe the adverse effects of commonly used hematology therapeutics for non-malignant blood disorders.
Drug: Epoetin Alpha (EPO)
Red blood cell (RBC) formation, also known as erythropoiesis, is stimulated by a hormone called erythropoietin. Erythropoietin is produced by kidneys when they detect low blood oxygen. Erythropoietin then stimulates bone marrow cells to increase RBC production. Any disorder that results in anemia will stimulate kidneys to produce erythropoietin.
What is a disorder where kidneys can’t produce erythropoietin that also results in anemia?
Answer: certain kidney diseases, particularly chronic kidney disease!
Of note, erythropoietin is primarily made in the liver prenatally. However, the production switches over to the kidneys, and kidneys are the primary source of erythropoietin in adults.
Health problems that result in anemia or suppress bone marrow production leading to anemia then cause the kidneys to sense low O2 carrying by red cells (due to lack of red cells or other red cell disorders where the cells aren’t carrying O2 – more later) and produce erythropoietin. There are many examples of such health problems that cause anemia: Infections, AIDS, bone marrow disorders, iron deficiency, etc.
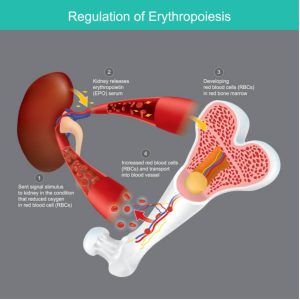
Epoetin Alfa (EPO) is a recombinant erythropoietin. It activates the JAK/STAT signaling pathway by binding to Erythropoietin receptors (Epo-R) on cells to stimulate red blood cell formation.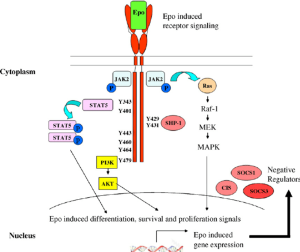
Epoetin alfa is given to patients with chronic kidney disease who have significant anemia due to lack of kidney production of erythropoietin. It can also be used to stimulate red cell production in Jehovah’s witness patients (who refuse blood products due to religious beliefs) to help correct more severe anemias in these patients. EPO can also be given to stimulate red blood cell production in the setting where red cell production is halted. In rare instances, EPO can be given to patients who have halted RBC production due to chemotherapy, but this has fallen out of favor due to some literature indicating worse cancer outcomes when EPO is used to treat chemotherapy-induced anemia.
Of note, Epoetin Alfa is NOT used to treat anemia due to sickle cell disease, as in that case the EPO would simply stimulate production of red cells that have Hemoglobin S, and Hemoglobin S is the problem with sickle cell disease. Similarly, this drug is not generally used in treating thalassemias.
Drug: Hydroxyurea
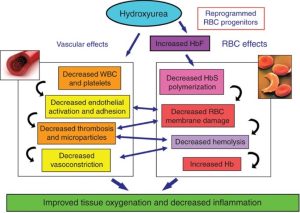
Contrasting with epoetin alfa, which stimulates RBC production, hydroxyurea is a drug that slows down bone marrow cell production. Hydroxyurea actually has 4 different mechanisms that are utilized in treatments of various hematological disorders.
- Hydroxyurea inhibits ribonucleotide reductase, which results in reduced bone marrow cell proliferation. This leads to decreased WBC, RBC, and platelet production.
- Hydroxyurea activates soluble guanylyl cyclase to increase fetal hemoglobin (HgF) gene expression. Increased fetal hemoglobin expression can be helpful to treat patients where the adult hemoglobin genes are mutated, such as thalassemias and sickle cell anemia.
- Hydroxyurea improves reticulocyte hydration, which can help with hemolysis in the setting of sickle cell disease.
- Hydroxyurea causes release of Nitric Oxide (NO) moiety, which leads to local vasodilation when NO is released. This can be helpful in the setting of vaso-occlusion during a sickle cell crisis and can offset the NO consumption that occurs with hemolysis.
Adverse effects of Hydroxyurea include bone marrow suppression resulting in cytopenias. Hydroxyurea can also cause cutaneous problems, including cutaneous and mucosal ulcers (leg/shin ulcers, for example) that can be dose-limiting.
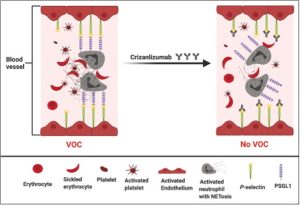
Drug: Crizanlizumab
Crizanlizumab is a humanized monoclonal antibody that has been developed against P-selectin. P-selectin is a molecule found on the surface of platelets and endothelial cells that line blood vessels. Normally, P-selectin binds with P-selectin glycoprotein ligand 1b-alpha (PSGL-1) on endothelial and white blood cells. Crizanlizumab blocks this interaction by binding to -selectin such that P-selectin can’t bind to PSGL-1. This prevents occlusion formation in blood vessels. In other words, this drug helps prevent vaso-occlusive crises (aka vaso-occlusive disease in patients with Sickle Cell disease. It is given as an injection once every 4 weeks. Side effects include nausea, joint pain, and fever.
Drug: L-glutamine
Supplementing L-glutamine increases the amount of free glutamine circulating in the blood. Glutamine is taken up by sickle cells and is used to generate antioxidants which then helps neutralize the oxidative stress causing increased stiffness of red blood cells in sickle cell disease. This allows sickle cells to regain flexibility needed to carry oxygen throughout the body.
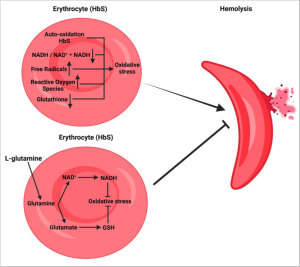
We have covered several drugs used to treat sickle cell disease: Hydroxyurea, Crizanlizumab, and L-glutamine. Here is some context on how these drugs are used to treat Sickle Cell Anemia:
- Hydroxyurea should be started in the first year of life at maximally tolerated doses.
- If hydroxyurea is not sufficient in reducing vasoocclusive events, then Crizanlizumab can be added to hydroxyurea as an additional treatment in patients 16 years of age or older.
- Additonal treatments for sickle cell crisis/vasoocclusive events include pain medication, hydration, and transfusion/exchange transfusion.
- L-glutamine is also FDA approved and may add marginal benefit in patients 5 years of age or older if the clinical response to Hydroxyurea is poor.
- If a suitable HLA-matched bone marrow donor is available, hematopoietic stem cell transplantation should be offered.
Drug: Filgrastim
In humans, white blood cell production by the bone marrow is accentuated by a growth factor called granulocyte-colony stimulating factor (G-CSF). G-CSF is produced by several cells including moncytes, macrophages, fibroblasts, endothelial cells, and bone marrow stromal cells. G-CSF binds to the G-CSF receptor on bone marrow precursor cells, leading to activation of the JAK/STAT pathway associated with the G-CSF receptor. Pathway activation stimulates the expression of genes that promote neutrophil proliferation and differentiation from bone marrow precursor cells.
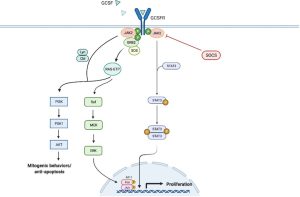
Filgrastim is a G-CSF analog. It binds to the G-CSF receptor (likek G-CSF) to promote neutrophil proliferation and differentiation. Filgrastim is used to treat neutropenia (lack of neutrophils) caused by chemotherapy. It can also be used to treat other forms of neutropenia such as some cases of autoimmune neutropenia. Filgrastim is also utilized to mobilize stem cells from the bone marrow of stem cell donors such that the stem cells will circulate into peripheral blood and can be harvested from stem cell donors by a process called apheresis and stored to be infused later into the recipient of the stem cell transplant.
Notice that both Epoeotin alpha (the first drug we discussed above) and filgrastim act on a receptor that involves JAK/STAT signalling!
Drug: Prednisone
Prednisone is a corticosteriod. Prednisone is generally used as an immune suppressant in various autoimmune diseases. Its mechanism of action is that it increases apoptosis of plasma cells, which are the blood cells that produce antibodies. (FYI, Plasma cells are derived from B-lymphocytes, and you’ll here more about this later. However, Prednisone is often involved in treating lymphocyte cancers such as lymphoma and lymphocytic leukemia. Prednisone also downregulates the phagocytic activity of macrophages. In an autoimmune disorder called Immune Thrombocytopenic Purpura or “ITP”, prednisone reduces production of antibodies targeting platelets, downregulates phagocytosis of platelets by macrophages, and also reduces production of platelet autoantigens. All of this leads to an increase in number of platelets in patients with ITP.
Drug: IV Immunoglobulins (IVIG)
IVIG is a blood product composed of pooled immunoglobulins from many people’s blood plasma. It can be given intravenously for various medical reasons. For example, it is used to treat some people with hypogammaglobluinemia (patients with low antibody levels) if those patients have been having repeated infections. In the setting of ITP (Immune Thrombocytopenic Purpura), IVIG has activity in treating this disease, which is not well understood. It is thought that IVIG may bind Fc receptors on macrophages and thus interfere with macrophages binding to the Fc portion of antibodies bound to platelets such that the macrophage does not phagocytose auto-antibody bound platelets in patients with ITP. Effects of IVIG are generally short-lived, and IVIG is often used in newly diagnosed ITP while other therapies are being arranged.
Drug: Thrombopoietin (TPO) Receptor Agonists: Romiplostim and Eltrombopag
These drugs both bind to and activat the thrombopoietin receptor. The thrombopoietin receptor is found on bone marrow cells such as megakaryocytes. TPO receptor agonists such as romiplostim and eltrombopag stimulate megakaryocyte survival and proliferation and, hence, increase platelet production. Would you care to guess what pathway is activated when these drugs stimulate the TPO receptor? If you guessed the JAK/STAT pathway, you would be correct!! So, yes, the drugs that stimulate bone marrow production of red blood cells, neutrophils/WBCs, and platelets all act on receptors that lead to activation of JAK/STAT pathways in those cells that have those receptors!
Romiplostim is an antibody-peptide fusion protein. It is given as an injection under the skin (subcutaneous injection) and binds the active site of the TPO receptor.
Eltrombopag is a small hydrazone molecule. It is given orally (taken by mouth) and binds the transmembrane portion of the TPO receptor.
Both drugs stimulate megakaryocyte survival and proliferation, leading to increased platelets. They are generally used as treatments for ITP, if the ITP is refractory to prednisone therapy (in other words, TPO receptor agonists are used as 2nd line therapy for ITP).
Eltrombopag (but NOT romiplostim) is also used to treat a bone marrow failure problem called aplastic anemia. It is thought that Eltrombopag acts on TPO-R expressed by hematopoietic stem cells and multipotential bone marrow progenitor cells (as well as maturing megakaryocytes).
Side effects of these drugs include thrombosis and, in the case of eltrombopag, hepatotoxicity.
Drug: Rituximab
Rituximab is a monclonal antibody that binds to CD-20 antigen, which is present on B lymphocytes (and also on CD-20 expressing B-lymphoma cells, which are cancerous B-lymphocytes). Recall that B-lymphocytes also give rise to plasma cells, and that plasma cells produce antibodies. Hence, Rituximab is used in several autoimmune disorders to suppress the immune system by destroying B-lymphocytes and reducing auto-antibody formation. As you can image, rituximab is another medication that can be used to treat refractory ITP. Common side effect of rituximab is an infusion reaction to this antibody. Rituximab is also used to treat CD-20 expressing hematopoietic cancers such as B-cell non-Hodgkin lymphoma. You’ll hear more about Rituximab (Rituxan) later in this course.
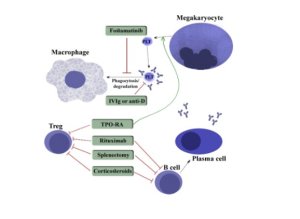
The diagram below outlines various treatments of ITP. We have covered prednisone (corticosteroids), IVIG, TPO-receptor agonists, and Rituximab. The other treatments outlined below are just FYI. Of note, synthetic Anti-D antibody is also called Win-Rho, and this an antibody used in Rh negative pregnant women to prevent them from developing anti-D antibodies if their fetus is Rh positive. You’ll learn more about Rh or D antigen of red blood cells in transfusion medicine. Interestingly, Win-Rho, the synthetic anti-D antibody, also can be used to treat ITP. Like IVIg, it probably interferes with macrophage recognition of anto antibody-coated platelets by otherwise occupying the Fc binding site on macrophages, etc. This is just FYI, in case you see this being done clinically.
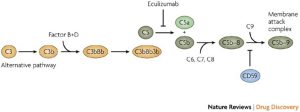
Drug: Eculizumab
Eculizumab is a monoclonal antibody that binds the complement cascade component C5. It inhibits conversion of C5 to C5a and C5b. Recall that the complement cascade is part of the innate immune system that you learned about in I&I. Basically, Eculizumab is a drug designed to shut down complement activation. It is used to treat a disorder called Paroxysmal Nocturnal Hemoglobinuria (PNH).
PNH is an acquired disorder that occurs when multipotent hematopoietic stem cells acquire a mutation in the PIGA gene, which is responsible for the first step in the synthesis of the glucosylphosphatidylinositol (GPI) anchor that attaches a subset of proteins to the cell surface. It is thought that the PIGA gene mutation somehow provides a conditional survival advantage to the PNH cell clone. However, the mutation also results in decreased synthesis of GPI anchors, which is a glycolipid that links dozens of cell-surface proteins to the plasma membrane on hematopoietic cells. This includes the ability to link complement regulatory proteins on the surface of cells. Hence, in the setting of PNH, hematopoietic cells such as red blood cells lack these complement regulatory proteins on their surface. Thus, these cells are much more sensitive to complement-mediated lysis. PNH disease is characterized by complement hemolysis as well as increased risk for blood clots and for impaired bone marrow function.
Eculizumab impairs complement activation via inhibition of activation of C5, and hence is used to treat PNH. Risks of Eculizumab include increased risk of life-threatening infections including meningococcal infections (think of infections that our immune system treats via the complement pathway).
Drug: Desmopressin (DDAVP)
Desmopressin is a vasopressin analog. It binds to vasopressin type 2 receptors (V2R) and stimulates release of von Willebrand factor from endothelial cells lining the blood vessel wall. In addition, it increases secretion of clotting factor VIII. It is used to treat certain types of von Willebrand disease.
Von Willebrand factor is a protein involved in stopping bleeding. Its job is to bind platelets to each other and to vascular endothelial cells, when the endothelium is disrupted, and help produce a platelet plug when vascular endothelial cells are damaged. VWF is in the circulation but is also stored in vascular endothelial cells. Patients who lack von Willebrand factor are prone to bleeding, and this condition is called von Willebrand Disease. Von Willebrand Disease is the most common inheritable bleeding disorder. There are several different types of Von Willebrand disease, as defects in von Willebrand factor can be quantitative (low or absent amounts of von Willebrand factor) or qualitative (decreased function of von Willebrand factor).
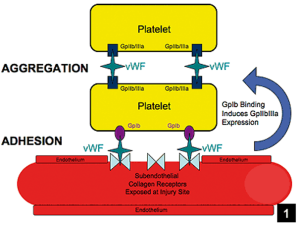
Type 1 von Willebrand Disease is an autosomal dominant disorder where there is decreased production of von Willebrand factor (VWF). VWF is produced, just in lower amounts. Desmopressin (DDAVP) is commonly used to stop bleeding in type 1 von Willebrand Disease, as it can cause release from endothelial cells of their stored VWF to stop bleeding in the acute setting.
Type 3 von Willebrand Disease is an autosomal recessive disorder with absent production of VWF. Obviously, DDAVP won’t work for treatment of type I von Willebrand Disease, as DDAVP only works by increasing release of VWF, which won’t happen if there isn’t any stored VWF to begin with!
Drug: Caplacizumab
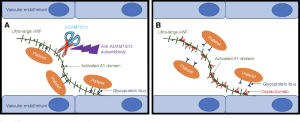
The von Willebrand factor (VWF) that we just mentioned above actually can circulate in blood in various sized multimers. Ultra-large multimers of VWF can be a problem if they accumulate on vascular endothelial cells and ensnare platelets, especially in areas of high shear stress such as small arterioles and capillaries. Our body has a way of preventing this in the form of ADAMTS13 protease (A Disintegrin And Metalloprotease with a ThromboSpondin type 1 motive, member 13) protease. ADAMTS13 protease is a von Willebrand factor-cleaving protease. It cleaves the ultra-large, string-like molecules of VWF that are synthesized by endothelial cells and secreted into plasma but remain attached to the endothelial surface. This normal cleavage of larger multimers of VWF by ADAMTS13 protease to smaller sized von Willebrand Factor multimers prevents ultra-large multimers from accumulating. Basically, in areas with high shear stress, the ultra-large multimers of VWF have a conformational change such that the ADAMTS13 cleavage site is exposed and ADAMTS13 protease can do it’s job and cleave the multimers. If these large multimers aren’t cleaved, then when they expand in areas of high shear stress, they trap platelets and clotting can ensue.
Acquired Thrombotic Thrombocytopenic Purpura (TTP) is a disorder where a patient has developed anti-ADAMTS13 protease antibodies. These antibodies against ADAMTS13 protease reduce ADAMTS13 protease activity and inhibit proteolytic cleavage of ultra-large VWF multimers. GP1b-alpha receptors on platelets bind to activated A1 domains on VWF multimers and the platelet aggregation can lead to microvascular thrombosis, resulting in life-threatening ischemic organ damage.
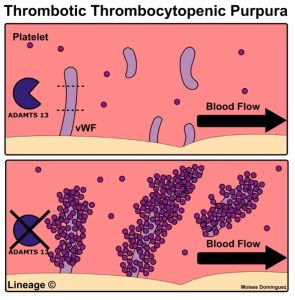
Caplacizumab is a humanized monoclonal antibody that binds the A1 domain of ultra-large VWF. Caplacizumab blocks the interaction of von Willebrand factor with glycoprotein 1b-alpha receptors on platelets. This reduces platelet aggregation and microvascular thrombi formation. It is used to treat acquired TTP. Side effects include mucocutaneous bleeding (nose bleeds, bleeding gums), headache, and fatigue.
Authors and Contributors:
Corliss Newman, MD wrote and Edith Wang, PhD edited this to correspond with the pharm lecture material prepared by Edith Wang, PhD.