
4 Sickle Cell Disease and Thalassemias
Learning Objectives
- Describe the inheritance of sickle cell disease.
- Explain the molecular basis for polymerization of deoxyhemoglobin S and the process of sickle vaso-occlusion.
- Recognize the clinical manifestations and treatment of sickle cell disease: acute pain crises and progressive organ failure.
- Recognize the molecular genetics and inheritance of alpha- and beta- thalassemias.
- Describe the clinical and laboratory features of Beta-thalassemia minor (trait) and Beta-thalassemia major and the different types of alpha-thalassemias.
Required Textbook Reading:
- Chapter 9 Sickle Cell Disease (Section on Diagnosis and Clinical Manifestations, Brief section on hydroxyurea therapy).
Optional Textbook Reading:
- Chapter 8 Thalassemia
- Chapter 9 Sickle Cell Disease (additional sections not included above)
The Structure of Hemoglobin
To understand Sickle Cell disease and Thalassemias, one first needs to understand the structure of hemoglobin. Normal Hemoglobin molecules are composed of 4 subunits: two alpha-like chains and two beta-like chains. One alpha and one beta chain each form a dimer and then the 2 dimers form a symmetric tetramer. Each globin chain provides a pocket for one heme-iron moiety. Hence, the final tetramer can bind four heme-iron moieties.
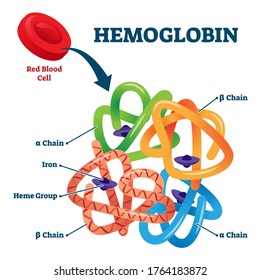
The beta-like globin chains, also called the beta-globin gene cluster, are encoded on chromosome 11.
The alpha-like globin chains, also called the alpha-globin gene cluster, are encoded on chromosome 16.
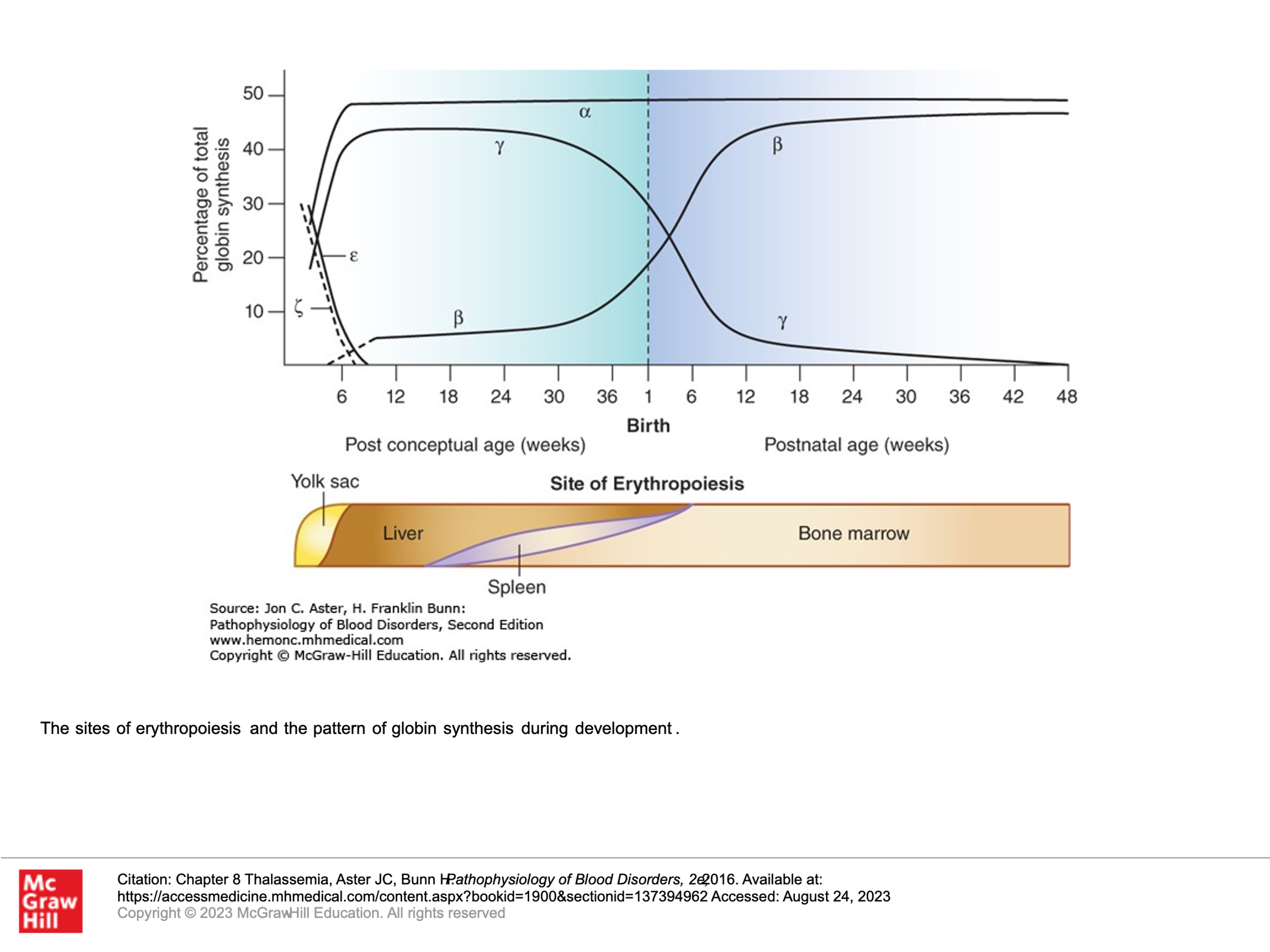
The beta-like and alpha-like globin genes are expressed at different times in development. The picture below demonstrates these genes as they appear on chromosomes 16 and 11, respectively.
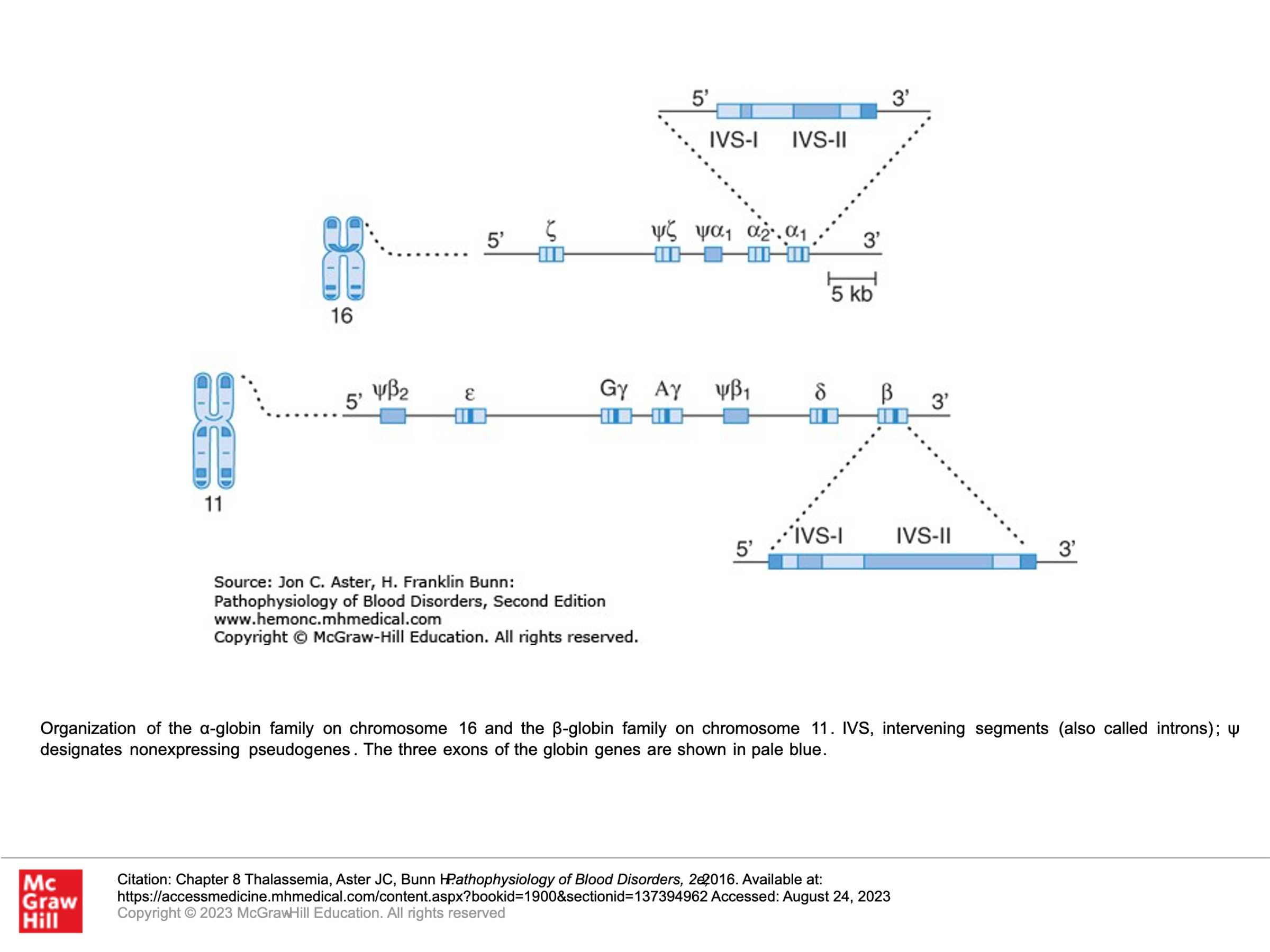
Alpha-like globin genes:
z: Zeta-globin (an early alpha-like globin) is expressed in the yolk sac prenatal stage of development and is gone (no longer expressed) by about week 8 gestation.
a and a: There are two alpha-globin genes on chromosome 16. These are expressed in the yolk sac prenatal stage of development and then becomes the dominant alpha-globin chain by about week 8 of gestation and persists into adulthood.
Beta-like globin genes:
e: Epsilon-globin is expressed in the yolk sac prenatal stage of development and is gone (no longer expressed) by about week 8 gestation.
g: Gamma-globin genes (there are 2 on chromosome 11) are expressed from the yolk sac stages of gestation and become the dominant Beta-like globin genes expressed thru birth. The expression of the g-globin genes then decreases and becomes negligible by about 24 weeks after delivery.
d: Delta-globin gene expression is very low level, starting at just before 30 weeks gestation and then persisting as about 3-4% of beta-like globin chain produced into adulthood.
b: Beta-globin gene expression starts at about week 8 of prenatal gestation and rises such that by birth it is slightly less than gamma-globin production. Beta-globin expression takes over as the dominant beta-like globin that is expressed by about 18 weeks after delivery.
Embryonic Hemoglobin:
z2e2
Post-embryonic Hemoglobin species:
a2g2 = Hemoglobin F (Fetal Hemoglobin, HgbF, most abundant fetal hemoglobin, but comprises <1% of adult hemoglobin)
a2d2 = Hemoglobin A2 (HbA2, <3.5% of adult hemoglobin)
a2b2 = Hemoglobin A (HbA, >95% of adult hemoglobin)
Hemoglobin Disorders (hemoglobinopathies)
Disorders in Hemoglobin production can be either Qualitative or Quantitative.
Qualitative Hemoglobin disorders: These are generally due to a genetic issue causing an amino acid substitution that results in a hemoglobin that has structural abnormalities. Examples: Hemoglobin S (sickle hemoglobin), Hemoglobin C, Hemoglobin E, Hemoglobin D. Note, in these disorders, there is normal ratio of alpha-like and beta-like proteins. Red cell production results in normal-sized red blood cells (normocytic) but the globin proteins can have structural problems that affect their function. Sickle cell disease is the most well-known qualitative hemoglobin disorder.
Quantitative Hemoglobin disorders = thalassemias: These types of gene mutations usually result in reduced or absent production of globin proteins (globin gene duplication, either alpha- or beta-globin genes, can also cause an imbalance in the abundance of alpha- and beta-globin proteins, also resulting in pathology). A deficiency in alpha- or beta-chains results in smaller-sized red blood cells (microcytic RBCs) in settings where there is significant deficiency of hemoglobin production.
- Alpha-thalassemia: reduced or no production of a-chains, with resulting excess of beta globin chains.
- Beta-thalassemia: reduced or no production of b-chains, with resulting excess of alpha globin chains.
Combination Hemoglobinopathies:
In this situation, patients have inherited both a tendency to a thalassemia and a structurally abnormal hemoglobin. For example, hemoglobin S/Beta-thalassemia.
Why do we have genetic selection for thalassemias and certain hemoglobinopathies?
Hemoglobinopathies can be commonly found throughout the world, mainly in populations that come from sub-Saharan Africa, Spanish-speaking regions in the Western Hemisphere (South America, Cuba, and Central America), Saudi Arabia, India, and Mediterranean countries such as Turkey, Greece, and Italy. Why is this? Great question. These disorders can be severe in homozygous states, so one would expect that selective evolution would have led to decreased frequency of these gene mutations. However, it turns out that heterozygotes for these types of disorders have a selective survival advantage where falciparum malaria is endemic. Hence, over evolution, these mutations were selected for, as they provided a survival benefit in settings where malaria is endemic. Because of this, hospitals in the USA screen all newborn babies for sickle cell disease.
Sickle Cell Disease
Sickle cell disease (SCD) is a qualitative hemoglobin disorder. SCD occurs when a patient is homozygous for a mutation in beta globin gene (both genes mutated) where hemoglobin Beta position 6 glutamic acid is switched to a valine (E6V). This causes production of hemoglobin S (a2s2) as opposed to hemoglobin A (a2b2) as the main form of hemoglobin. This finding is not concerning when hemoglobin is bound to O2, as the conformation of the hemoglobin in that setting doesn’t promote polymerization. In the setting of hypoxia/acidosis, hemoglobin would normally not polymerize either. However, when valine is present at position 6, it is relatively hydrophobic. This position is exposed in settings of hypoxia/acidosis, and due to hydrophobic issues, the hemoglobin with a valine at position 6 tends to polymerize. This polymerization of hemoglobin in this setting causes the red cells to change shape into sickle shapes and other shapes that are more rigid, and this leads to hemolysis of the cells. In addition, the cells, being more rigid, can plug capillaries, and they also tend to stick to endothelial walls causing vaso-occlusion and ischemia/pain.
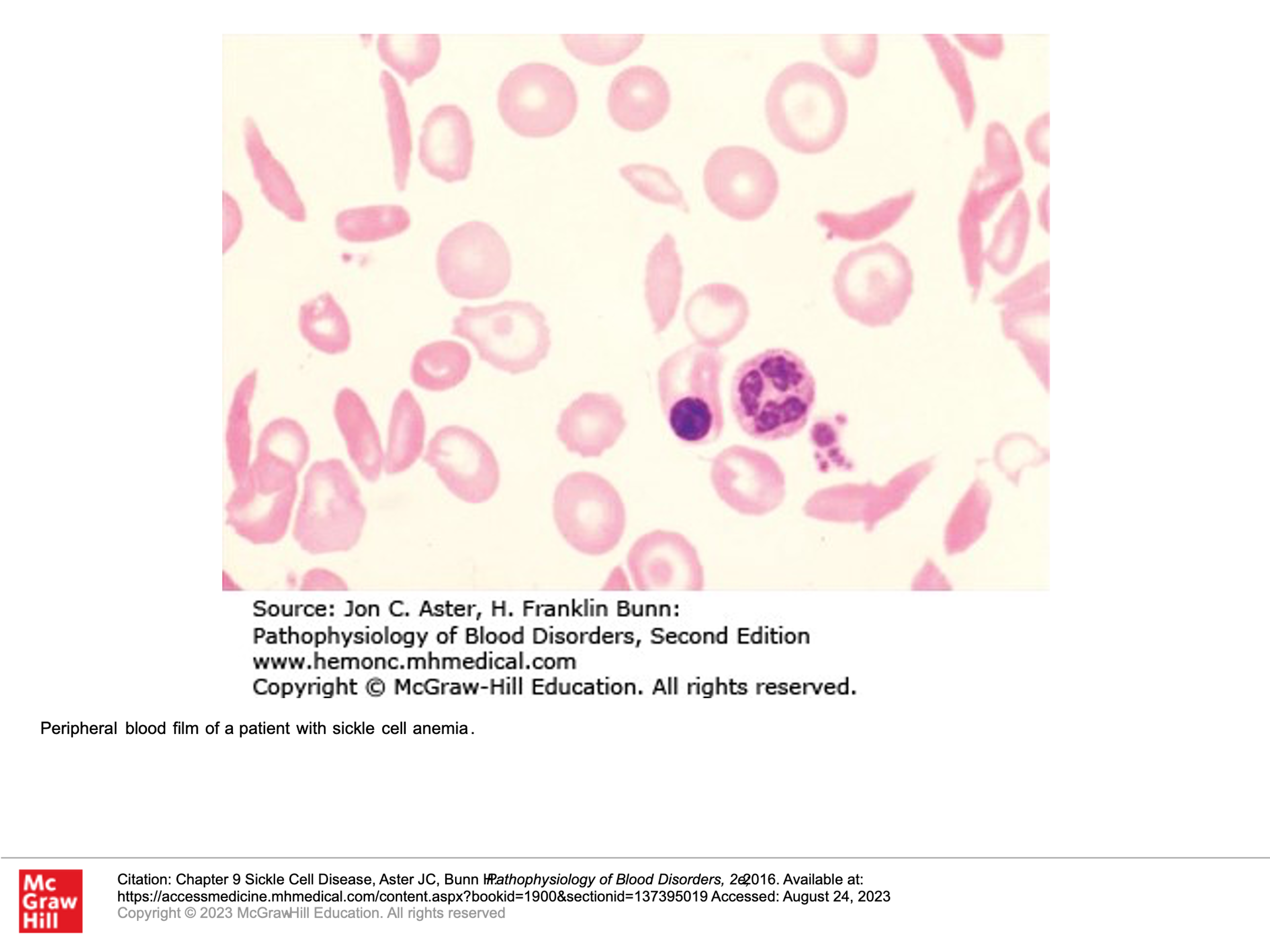
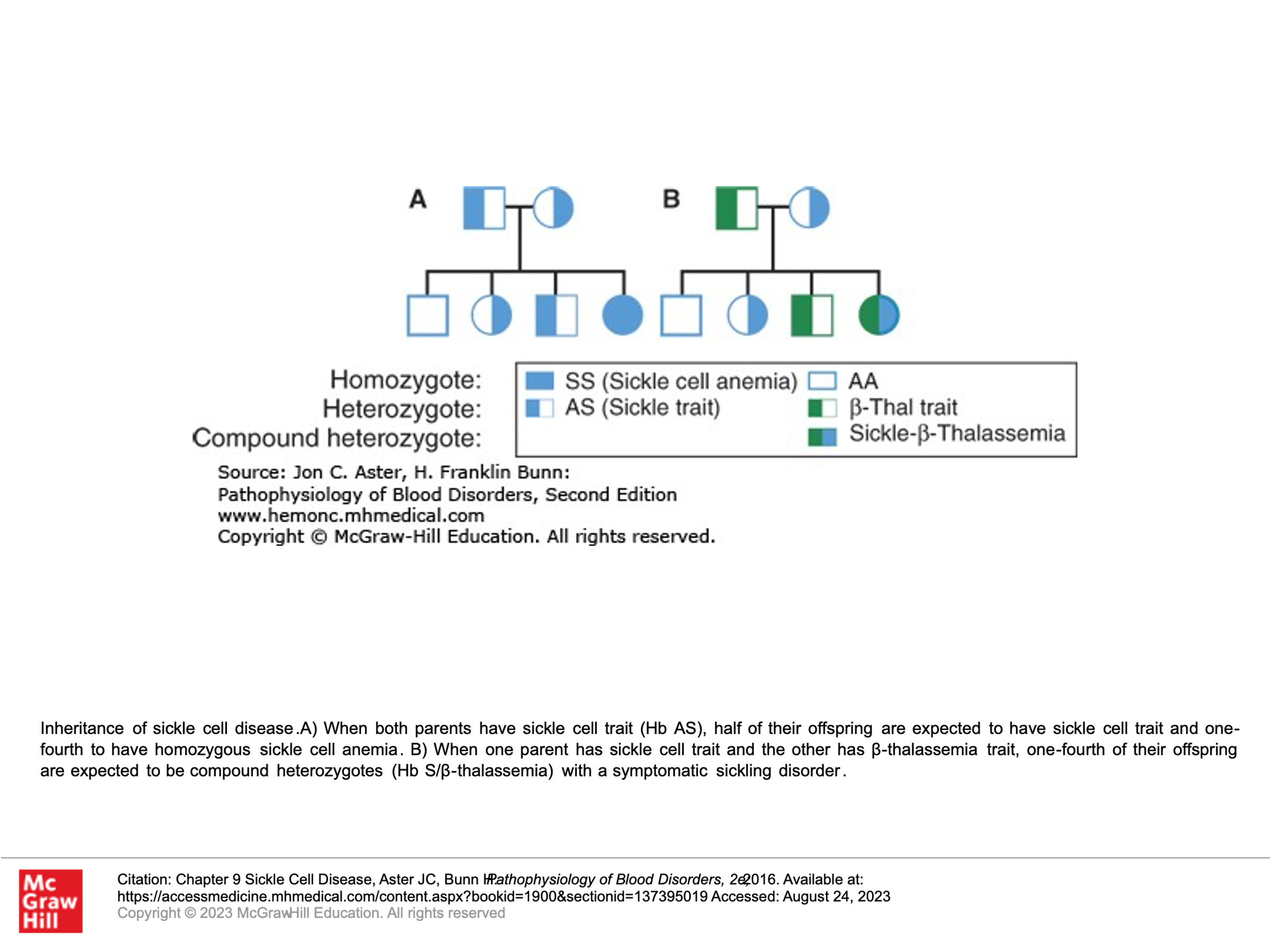
Inheritance of Sickle Cell disease:
Patients with sickle cell disease are generally homozygous for the beta-globin mutation that will give rise to hemoglobin S. They don’t have any hemoglobin A.
Patients with sickle cell trait are generally heterozygous for the beta-globin mutation that gives rise to hemoglobin S, such that they have both hemoglobin A and hemoglobin S.
Some patients are compound heterozygous, having both a beta-globin mutation that gives rise to hemoglobin S as well as another beta-globin mutation (commonly some type of Beta-thalassemia).
Complications of Sickle Cell disease:
Acute complications of Sickle cell disease include vaso-occlusion (pain), dactylitis (sausage-digit, inflammation of an entire digit), acute chest syndrome (fever, cough, chest pain, shortness of breath, low O2 levels, and increased sputum production), stroke, acute priapism, hepatic/splenic sequestration of RBCs, splenic infarcts, aplastic crisis in the setting of Parvovirus B19 infection.
Chronic complications of Sickle cell disease include heart failure, pulmonary hypertension, proliferative retinopathy, avascular necrosis, osteoporosis, leg ulcers, chronic kidney disease, and gall stones. Most children with sickle cell disease are functionally asplenic by age 6 years old, and thus would be at higher risk for infections from encapsulated organisms: Streptococcus pneumoniae, Neisseria meningitides, Klebsiella pneumoniae, Haemophilus influenza.
Additional key points about sickle cell disease:
Childhood mortality has dramatically improved with PCN prophylaxis in patients with sickle cell disease. Children with sickle cell disease are given Penicillin (PCN) prophylaxis until at least age 5, and this is often continued beyond age 5 in kids who have had their spleen removed or a serious pneumococcal infection.
Patients and their families/caregivers of people with sickle cell disease should be aware and reminded that fever (fever greater than or equal to 38.5 C or 101.3 F) in a patient with sickle cell disease is a medical emergency and patient should be seen immediately by a health care professional, regardless of whether the patient is taking prophylactic PCN.
Sickle Cell trait:
Sickle cell trait occurs, as shown above, when a patient is heterozygous for hemoglobin S and a normal beta- globin, so they have a mix of hemoglobin A and hemoglobin S. Such patients tend to have about 35-40% HbS and about 55-60% Hb A. These patients don’t generally have problems with sickling, but can have higher risks for some problems, including proteinuria/chronic kidney disease, pulmonary embolism, and exertional rhabdomyolysis.
How to Treat Sickle Cell disease in both the acute and chronic setting:
The hallmarks of treatment for patients with sickle cell disease include prevention of sickle cell crisis, management of acute and chronic complications of sickle cell disease, and consideration of potential cures.
Prevention of complications/crises:
- Routine health management with a health care provider with expertise in Sickle Cell disease.
- Use of PCN prophylaxis
- Keep up to date with immunizations.
- Consider blood transfusions for those at risk for stroke as a preventative measure.
- Use of Hydroxyurea to reduce the instance of acute vaso-occlusive pain episodes and other vaso-occlusive events.
Treatment of acute complications:
- Keep blood flowing: Give IV fluids, prevent dehydration. Ambulate. Give thromboembolism prophylaxis (DVT prophylaxis), Keep patients warm.
- Give antibiotics for infections.
- Optimize O2 delivery: Use incentive spirometry to improve oxygenation. Treat obstructive sleep apnea and asthma. Control pain so that patients will take deeper breaths, etc. Give O2 if hypoxic.
- Decrease presence of Hb S: Red cell transfusions, Red cell exchange transfusions. Treatment with Hydroxyurea promotes production of hemoglobin F (HbF, fetal hemoglobin).
- Medications to treat sickle cell disease:
- Crizanlizumab (monoclonal ab therapy): monoclonal antibody directed against p-selectin, a cell adhesion molecule involved in recruitment of leukocytes to sites of inflammation. upregulation of P-selectin in endothelial cells and platelets contributes to the cell–cell interactions that are involved in the pathogenesis of vaso-occlusion and sickle cell–related pain crises.
- Hydroxyurea: increases HbF and promotes nitric oxide production by an uncharacterized mechanism.
- Folic acid: A supplement needed due to hemolysis, which consumes folate.
- L-glutamine: This restores nitric oxide and improves oxidative stress.
- Gene therapy also being utilized now to treat sickle cell disease.
Potential Cures for Sickle Cell disease:
- Gene therapy is being explored.
- Bone marrow stem cell transplant is a known cure, but risks are significant.
Thalassemias
There are an inherited group of disorders in which mutations in globin genes result in impaired hemoglobin synthesis. This can result in anemia of varying severity and varying degrees (or none) of microcytosis. The thalassemias are divided into alpha or beta according to which globin genes are defective.
Alpha thalassemias vary depending upon how many alpha genes are disrupted (recall that each individual has four alpha genes because chromosome 16 carries 2 alpha genes from each parent). We’ll have more on this later below:
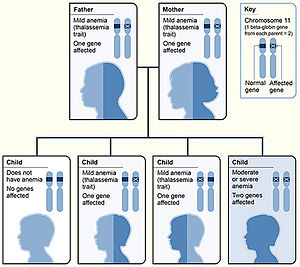
Beta Thalassemias:
Beta thalassemias have evolved some different nomenclature, as there are only 2 genes in every person, so clinical manifestations of having 1 or both beta-genes affected are generally more apparent/less variable:
b0 (Null): No gene product is produced from the mutated Beta gene (absent Beta-globin production).
b+: Beta gene has a mutation that results in reduced production of a Beta globin protein.
Beta thalassemias are classified clinically based upon clinical phenotypes:
Beta Thalassemia Minor (AKA Beta Thalassemia Trait): Common genotype is b0/b or b+/b. Patients are generally asymptomatic with a mild microcytic anemia with a hemoglobin generally >10g/dL. Hemoglobin electrophoresis will generally show elevated percentage of HbA2 and possibly mildly elevated HbF.
Beta Thalassemia Intermedia: Common genotype is homozygous or compound heterozygous such as b+/b+, b0/b+, HbE/b0, HbE/b+, others. Patients generally have a mild to moderate microcytic anemia, with a Hb of 7-10. They have variable severity with regards to other problems such as bone disease, iron overload, splenomegaly, or pulmonary hypertension. Hemoglobin electrophoresis will show elevated HbA2 (can be similar to Thal Minor) and elevated HbF (usually > 10% HbF).
Beta Thalassemia Major: Common Genotype is Compound Heterozygous or Homozygous: b0b0, b0b+, HbE/b0. Patients generally have severe anemia with a Hb less than 7 before age 2. They are generally transfusion dependent. Hemoglobin electrophoresis (before transfusion) will show elevated HbA2 and elevated HbF.
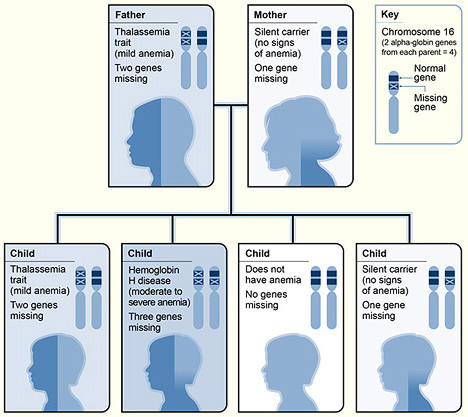
Alpha-Thalassemias:
As mentioned earlier, we all have 4 alpha genes. Hence, there are more variations of alpha thalassemias. Note that unlike the situation with beta-like chains (we have both beta and delta beta-like chains produced in adulthood—leading to HbA and HbA2), both HbA and HbA2 have alpha genes.
Normal: Person has all 4 alpha genes, no alpha genes are mutated.
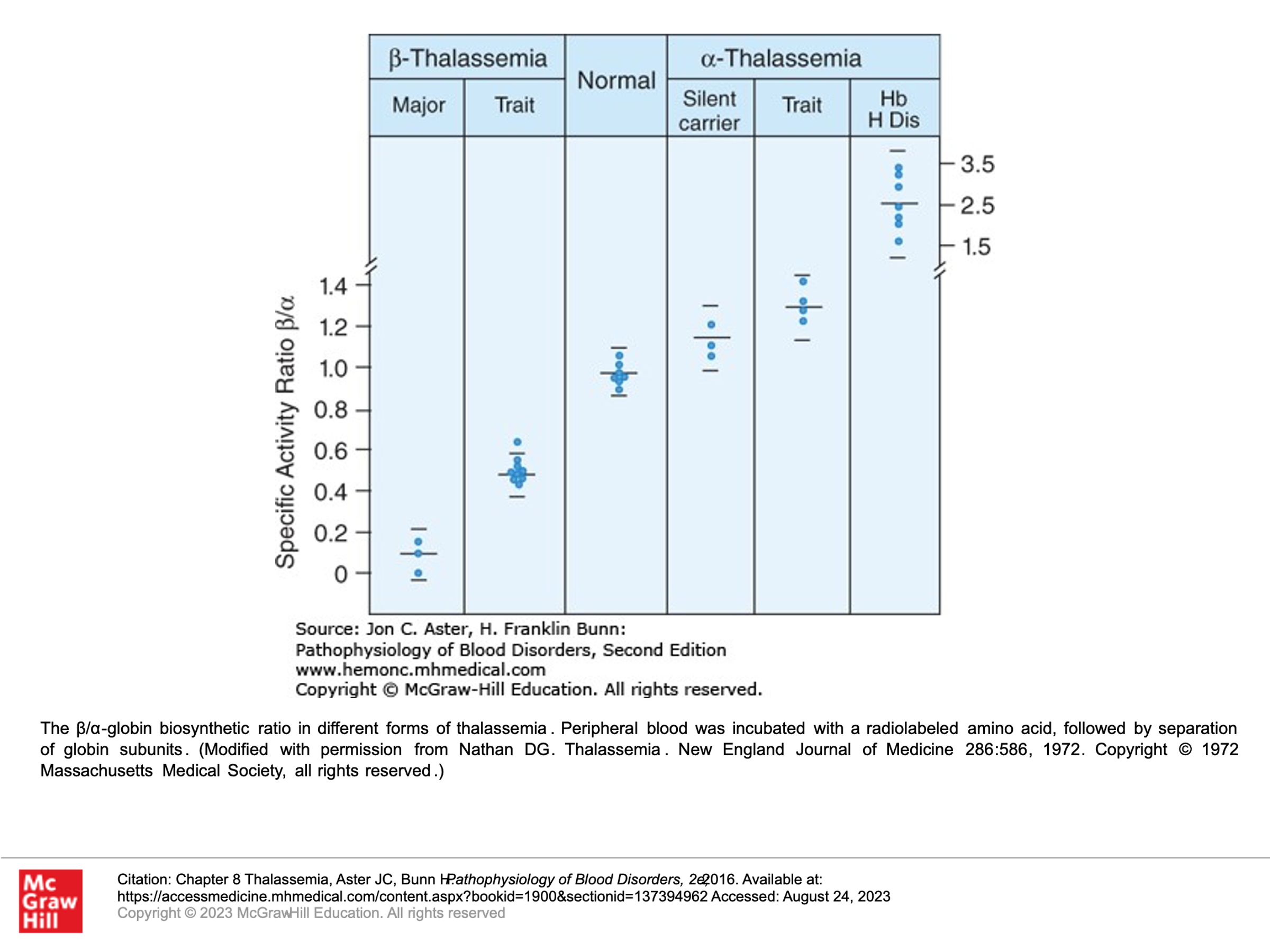
Carrier: Person has 3 out of 4 alpha genes normal, and 1 alpha gene is mutated. Generally, carriers are asymptomatic and have no CBC abnormalities.
Alpha-thalassemia Minor: Person has 2 of 4 genes normal, 2 alpha genes mutated. Note that these can either be both genes on one chromosome 16, or one of the 2 genes on each chromosome 16. Two alpha genes that are normal on one chromosome and both mutated on the other is called a cis mutation (aa/–). When one alpha on each chromosome is mutated, that is called a trans mutation (a-/a-). In both settings, patients generally have a mild microcytic anemia.
Hemoglobin H disease: Person has 1 out of 4 alpha genes normal, and 3 alpha genes are mutated. These patients generally have symptomatic hemolytic and microcytic anemia, as well as splenomegaly. About 10% of hemoglobin in their blood will be hemoglobin H (HbH, a tetramer of 4 beta-globin chains) after they reach full mature hemoglobin production.
Hydrops Fetalis: All 4 alpha-globin genes are missing: This is incompatible with life, as the globin present in such situations is all comprised of homotetramers of beta-like chains either 4 gamma chains (Hb Bart) or 4 beta chains (HbH). For hemoglobin to transport oxygen efficiently, it must be a heterodimer composed of two pairs of alpha-like/beta-like subunits. Homotetramers such as hemoglobin H (b4) or hemoglobin Bart (g4) have a very high affinity for oxygen and lack the subunit cooperativity such that they don’t effectively release O2 to tissues. Hence, even though the hemoglobin of a fetus with –/– alpha thalassemia is fully oxygenated, its release of O2 to tissues is markedly impaired, and the baby suffers from severe tissue hypoxia resulting in stillbirth.
Pathophysiology and complications of Thalassemias:
An impaired ratio of alpha to beta globin leads to red cell production problems/red cell pathology with ineffective erythropoiesis, which in turn leads to iron overload and anemia. Note, these patients can develop iron overload even in the absence of blood transfusions. Ineffective erythropoiesis leads to increased intestinal iron absorption by an incompletely understood mechanism, per uptodate.com. Iron overload is associated with iron deposition in tissues causing diabetes mellitus, growth deficiency, hypothyroidism, hypoparathyroidism, hypogonadism, liver disease/hepatic cancer, and renal disease. At the same time, malformed red cells are more likely destroyed in the spleen (hemolysis) leading to problems such as increased gall stone formation. Iron overload and hemolysis both are associated with a hypercoagulable state leading to leg ulcers, thrombotic events, and pulmonary hypertension. Anemia is associated with decreased tissue oxygenation and expansion of the erythroid marrow production. This expansion of bone marrow production can lead to bone deformities, osteoporosis, as well as drive extramedullary red cell production—causing hepatosplenomegaly and extramedullary hematopoietic cell deposits elsewhere that can mimic tumors (pseudotumors).
Treatment of Beta-Thalassemia major:
- Genetic counseling (including prenatal diagnosis).
- Red cell transfusions every 3 to 4 weeks to keep hemoglobin above 9.5 (this will help prevent extramedullary hematopoiesis, etc).
- Iron chelation to prevent manifestations of iron overload.
- Luspatercept: This is a SQ infusion given every 3 weeks that improves red cell maturation.
- Allogeneic stem cell transplant can be curative.
- Gene therapy can be curative and was approved in 2019 in the European Union using a product called betibeglogene autotemcel, a therapy containing approximately 24-400 million autologous CD34+ cells transduced with a lentiviral vector carrying a normally functioning beta-globin gene variant (you do not need to know this specific product). It can be used in patients with certain types of beta-thalassemia, but doesn’t work in all types. In 2022, the USA FDA also approved this therapy.
Treatment of alpha thalassemia:
This depends on the severity, but is the same as that for Beta-thalassemia Major, as above. Generally, Hemoglobin H disease is like Beta Thal Major, and alpha-thal minor is like beta thal intermedia or beta thal minor.
Combinations of Sickle Cell Trait and Thalassemia or other Hemoglobin Variants
Given that both Sickle cell trait and Thalassemias are selected for in settings endemic for malaria, one can predict that there are going to be people who inherit combinations of both thalassemia and sickle trait.
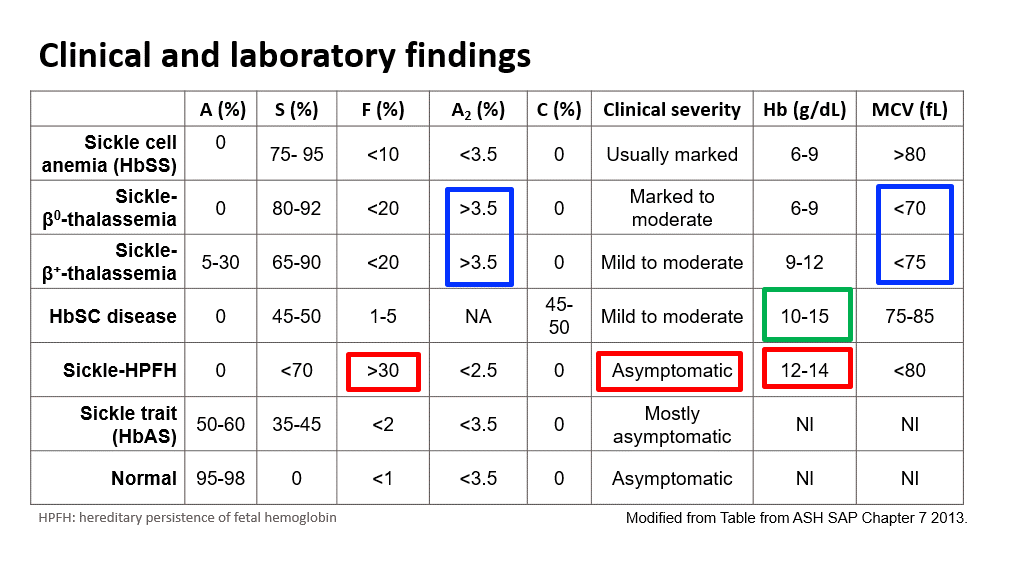
Here is a summary slide showing such combinations and comparisons between sickle cell disease vs various permutations of Sickle trait combinations: Note the percentages of hemoglobin A, F, A2, and S found in these different types of disorders with hemoglobin electrophoresis testing. Also note that only the disorders with a concurrent thalassemia have microcytosis. (Sickle cell disease and Sickle cell trait patients have CBCs with a normal MCV!) HPFH stands for Hereditary persistence of fetal hemoglobin. Hemoglobin C disease is mentioned in the text book (chapter on sickle cell disease), but basically Hb C is another beta-globin structural mutant (recall that Hb S is also a beta-globin structural mutant). Compound heterozygous patients with beta-S and beta-C alleles are designated as HbSC. This combination of allelic variants is also considered a sickle cell anemia, although much less severe than HbSS. Hemoglobin C disease (HbCC) is not a sickle cell anemia. In HbCC there is chronic hemolysis but not vaso-oclusion as HbC is not prone to polymerization. On peripheral smear you can see crystal shaped RBCs. This apperance reflects dense rectangular structures composed of precipitated hemoglobin C.
Also, note that in a standard hemoglobin electrophoresis, Hemoglobin A2 (alpha 2, delta 2) and hemoglobin C migrate the same. One must use an acid hemoglobin electrophoresis on citrate agar to further separate out Hb A2 and HbC.
Testing for Thalassemias and Sickle Cell Disease or other Hemoglobinopathies
Testing for hemoglobinopathies generally involves a test called hemoglobin electrophoresis. In a standard native gel hemoglobin electrophoresis, a drop of prepared blood is placed in a gel and then an electrical current is applied. Intact hemoglobin tetramers of different sizes (A, A2, S, C, etc.) will separate out on a gel due to the electrical current and can then be further identified. Other methods include high-pressure liquid chromatography (HPLC), capillary zone electrophoresis (CAE), and isoelectric focusing (IEF). You do not need to know these different tests. However, generally when one is concerned for thalassemia or sickle cell disease, a blood smear and a hemoglobin electrophoresis test is ordered.
Below and on the next couple pages are some examples/graphics of hemoglobin electrophoresis:
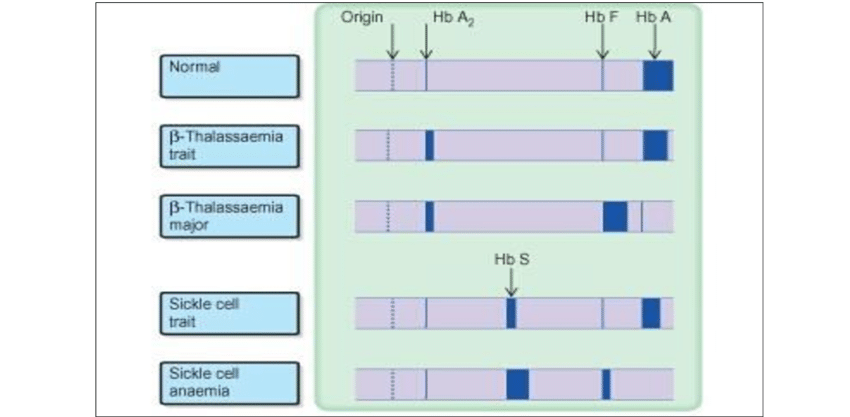
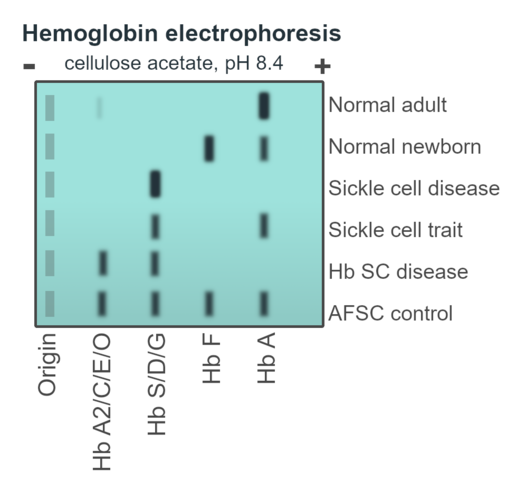
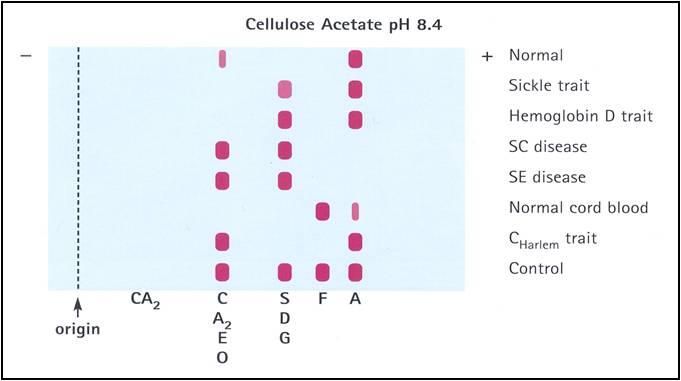
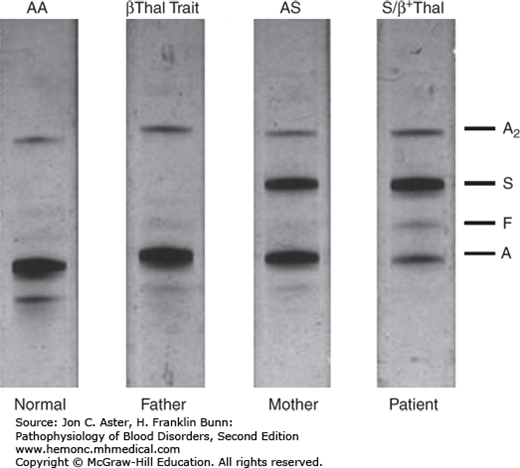
The normal Hb AA individual has predominantly HbA and 2% HbA2. The father has an increased HbA2 level (3.5%), indicative of β-thalassemia trait. The mother has nearly equal amounts of Hb A and Hb S, indicative of sickle cell trait. The patient inherited the β-thalassemia gene from his father and the βS gene from his mother. He has predominantly Hb S. The presence of a small amount of Hb A indicates that he has Hb S/β+-thalassemia. Note that the patient also has a slightly increased Hb F level.
Authors and Contributors:
Corliss Newman, MD authored this course pack material based partly upon slide lecture material prepared by Kleber Fertrin, MD PhD. Kristi Rice, MD and Nicholas Burwick, MD provided editing assistance.