
5 Overview of Hemostasis and Platelets
Student Learning Objectives:
- Recognize normal hemostasis, including the concepts of the coagulation cascade and the cell-based model of coagulation and primary and secondary hemostasis.
- Identify the mechanisms that limit blood clot formation.
- Identify the common causes and mechanisms of thrombocytopenia (congenital, production problem, destruction problem, sequestration).
- Describe examples of congenital and acquired platelet function disorders.
- Identify common inherited and acquired bleeding disorders.
UMSLE Content Descriptors: cytopenias, hypercoagulable, hypocoagulable, congenital disorders.
Optional textbook reading: Part II Hemostasis and Thrombosis, Chapter 13 (Overview of Hemostasis, sections on Platelet Adhesion and Activation and the Blood Coagulation Cascade)
Hemostasis
Hemostasis, or the clotting of blood/stoppage of bleeding, requires a primary process (primary hemostasis) whereby platelets plug holes and a secondary process (secondary hemostasis) by which the clotting cascade is activated to cause blood to stop flowing/clot. Once clotting has been achieved, the body then has a system to shut down clotting and to dissolve clots that have formed through a process called fibrinolysis.
Primary Hemostasis
Primary Hemostasis is the process by which platelets plug holes that have developed due to vascular damage. In this setting, with vascular damage, subendothelium is exposed to blood. Von Willebrand Factor (VWF) binds to subendothelial collagen and then binds platelets via the platelet GpIb (note that the I is a roman number 1 in this case) receptor causing the platelets to adhere to the area of subendothelial damage.
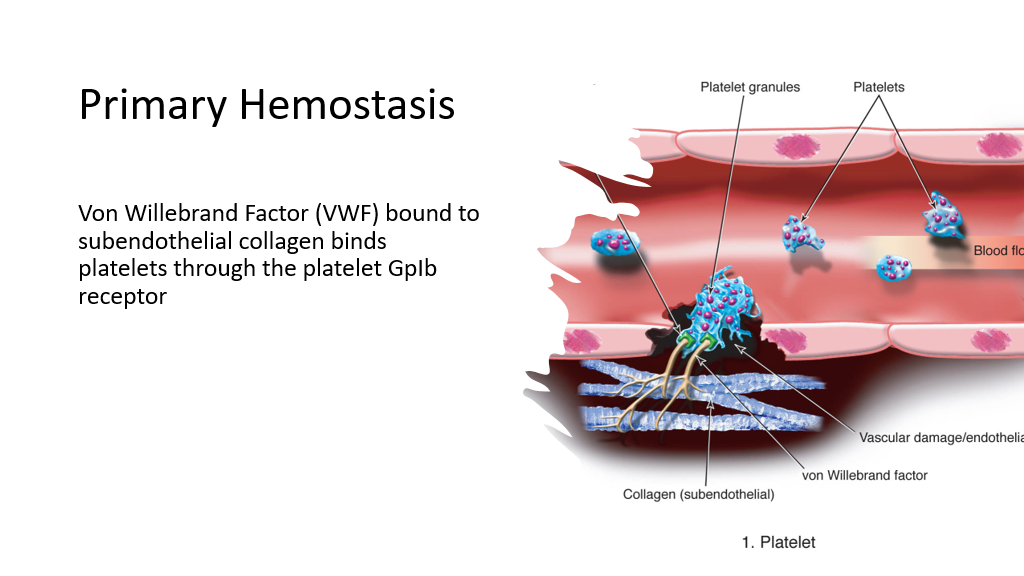
(Pausing here, can you think of situations where bleeding problems might occur? Yes, anything that affects this platelet plugging hemostasis process can result in bleeding tendencies: low platelets, lack of VW factor, defective VW factor, mutated GpIb binding site, etc. Nice job, you are well on your way to being a hematologist! Disorders that affect platelets and platelet-binding are considered disorders of primary hemostasis.)
Disorders of primary hemostasis generally present clinically with easy bruising, epistaxis, oral bleeding, GI bleeding, and menorrhagia.
Secondary Hemostasis
Secondary Hemostasis involves activation of the clotting cascade to further halt bleeding. When platelets bind to VW Factor/provide a platelet plug, the platelets are also activated. Activated platelets provide a phospholipid surface to support coagulation. Blood clot formation and its stabilization on the phospholipid surface is called secondary hemostasis. The ultimate goal of activation of the clotting cascade is the formation of fibrin, which will stop bleeding. Also, platelet granule release results in more local platelet activation. As part of the clotting cascade, multiple clotting factors are activated that result in formation of fibrin and crosslinking of fibrin.
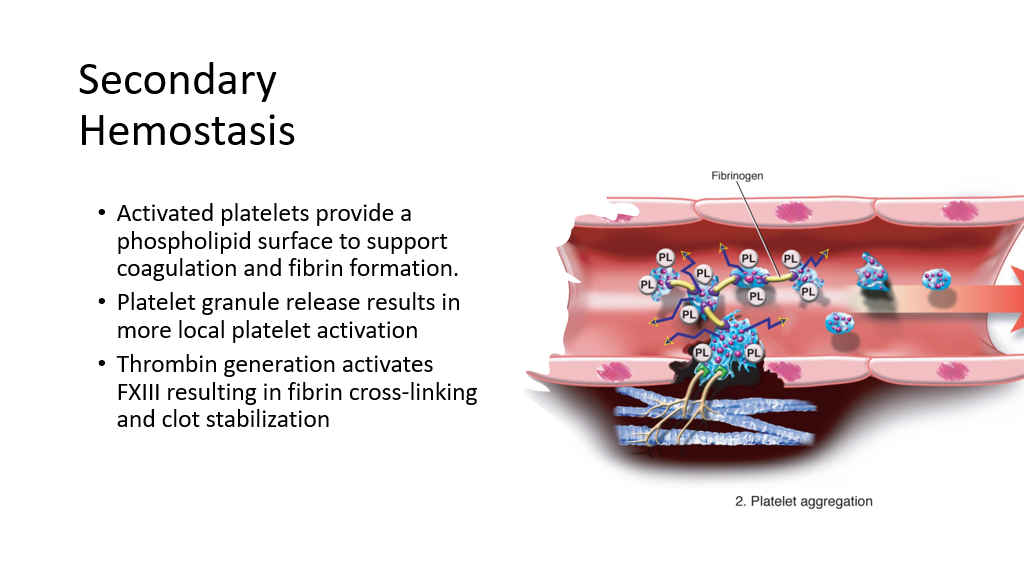
The Clotting Cascade
We are now going to go over the blood clotting cascade. As you learn the steps of this cascade, keep in mind situations that can result in bleeding or clotting, particularly in patients with disorders that affect the clotting cascade. Disorders of secondary hemostasis include deficiencies of blood clotting factors either due to inherited or acquired reasons. Disorders of secondary hemostasis tend to present with a slightly different pattern of bleeding than disorders of primary hemostasis. Patients with secondary hemostasis disorders can present with muscle and joint bleeding.
The clotting cascade was first described by Dr. Davie (a UW faculty member!) and Dr. Ratnoff in 1964. The clotting cascade is a series of clotting factors that are activated in what appears to be a cascading effect to rapidly ramp up clotting in the setting of bleeding. This cascade is composed of two types of clotting factors:
- Zymogens: These are clotting factors that are proenzymes that are then activated during hemostasis.
- Cofactors: These are clotting factors that are involved in steps in the clotting cascade by which zymogens are activated.
The clotting cascade ultimately leads to fibrin formation and stabilization via cross-linking, and resultant clot stabilization.
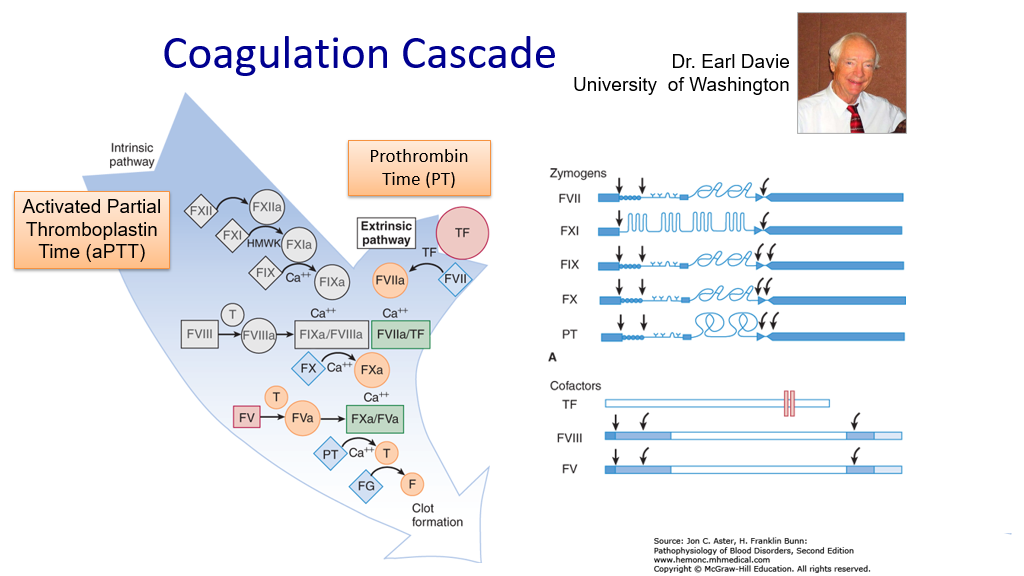
There are generally 2 pathways (yes, nothing is simple in medicine) that are considered in the clotting pathway: Both are measured by in vitro testing. These 2 pathways are as follows:
- The Intrinsic pathway (which is generally measured by the Activated Partial Thromboplastin Time, aka aPTT, aka PTT).
- The Extrinsic pathway (which is generally measured by the Prothrombin Time, aka PT—and remember that the PT is expressed in a normalized ratio called the INR—more on this later).
However, in vivo (in real life), things are a bit more complicated. To detect problems with bleeding, we need to be able to measure it, so we first talk about the pathways as measured via in vitro/lab testing (the PTT and PT).
Intrinsic pathway (measured by the Partial Thromboplastin time, PTT):
In the test tube, surface contact with the wall of the glass test tube triggers the intrinsic pathway. Exposure of the plasma to the glass surface of the tube triggers the activation of factor XII. Factor XII (a zymogen) is converted to its activated form (XIIa). XIIa then activates Factor XI (another zymogen) along with cofactor high-molecular-weight kininogen (HMWK). XIa (activated factor XI), in the presence of calcium ions, catalyzes the activation of factor IX to IXa.
Meanwhile, Factor VIII is activated to VIIIa via action of Thrombin (more on Thrombin later).
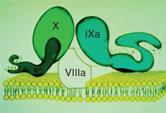
Factor IXa forms a complex with VIIIa in the presence of phospholipids and calcium ions to trigger the activation of factor X to Xa.
Meanwhile, Factor V is activated to Va via action of Thrombin (again, more on Thrombin later).
Xa and Va then form a complex, XaVa, which, in the presence of calcium ions, converts prothrombin to thrombin. (Note, “Prothrombin” is labeled as “PT” in the picture below, which is not the same as prothrombin time-which we also tend to abbreviate PT, so don’t get confused here.) Prothrombin is also called factor II, and, hence, Thrombin is also called activated factor II or IIa.
Thrombin then works to convert Fibrinogen to Fibrin, leading to clot formation.
Extrinsic pathway (measured by the Prothrombin time, PT):
Tissue factor (TF, which is also sometimes called Factor III) is added to the test tube and activates Factor VII to VIIa (activated factor VII). VIIa then complexes with tissue factorand calcium to further convert factor X to Xa. As before, Xa complexes with Va to then activate Prothrombin to Thrombin. Thrombin then converts inactive fibrinogen to Fibrin, to promote the formation of the Fibrin clot. Again, don’t forget that Factor XIII crosslinks the fibrin to make it more stable/secures the clot.
A side note on the INR: As you may know, most labs focus, for comparison purposes, on the INR (International Normalized Ratio) result and not the PT. This is because the prothrombin time will vary across labs due to variations between the different types and batches of tissue factor used in the testing process. The INR was devised to standardize the PT results so that they can be compared across institutions, etc. (obviously very important for monitoring therapeutic levels of the anticoagulant, warfarin). Each manufacturer of Tissue factor assigns an ISI value (International Sensitivity Index) for any tissue factor they manufacture. The ISI value indicates how a particular batch of tissue factor compares to an international reference tissue factor. The INR is the ratio of a patient’s prothrombin time to a normal (control) sample, raised to the power of the ISI value for the analytical system being used by that lab.
Common pathway: Note, both the Intrinsic and Extrinsic pathways converge at the formation of Xa. Hence, Activation of factor X and beyond is called the common pathway.
Question: If the PT is prolonged and the PTT isn’t in a patient with a bleeding disorder due to a factor deficiency, then what factor has to be the one that is deficient? Hint, it’s not tissue factor….Tissue factor is present in subendothelial tissues and leukocytes, and there is not a known viable deficiency of this protein. Answer: Factor VII
Honorable mention of Factor XIII:
Factor XIII (unlucky factor XIII never gets any credit and doesn’t get shown in pictures of the clotting cascade, as it is NOT measured by the PTT or the PT) is activated around this time to crosslink the fibrin to make it more stable. Does a lack of XIII lead to bleeding—yes! Lack of factor XIII results in delayed bleeding AFTER the clot first forms, as the clot is not stabilized by fibrin crosslinking, and thus the clot breaks down more easily! Again, this won’t be measured by the PTT or PT, or another test called the thrombin time (more on this later). (If you are interested, Factor XIII deficiency can be found with an abnormal Urea clot lysis test, but this is beyond the scope of this course.)
The clotting cascade in vitro (what happens in the test tubes when we measure the PTT and PT):
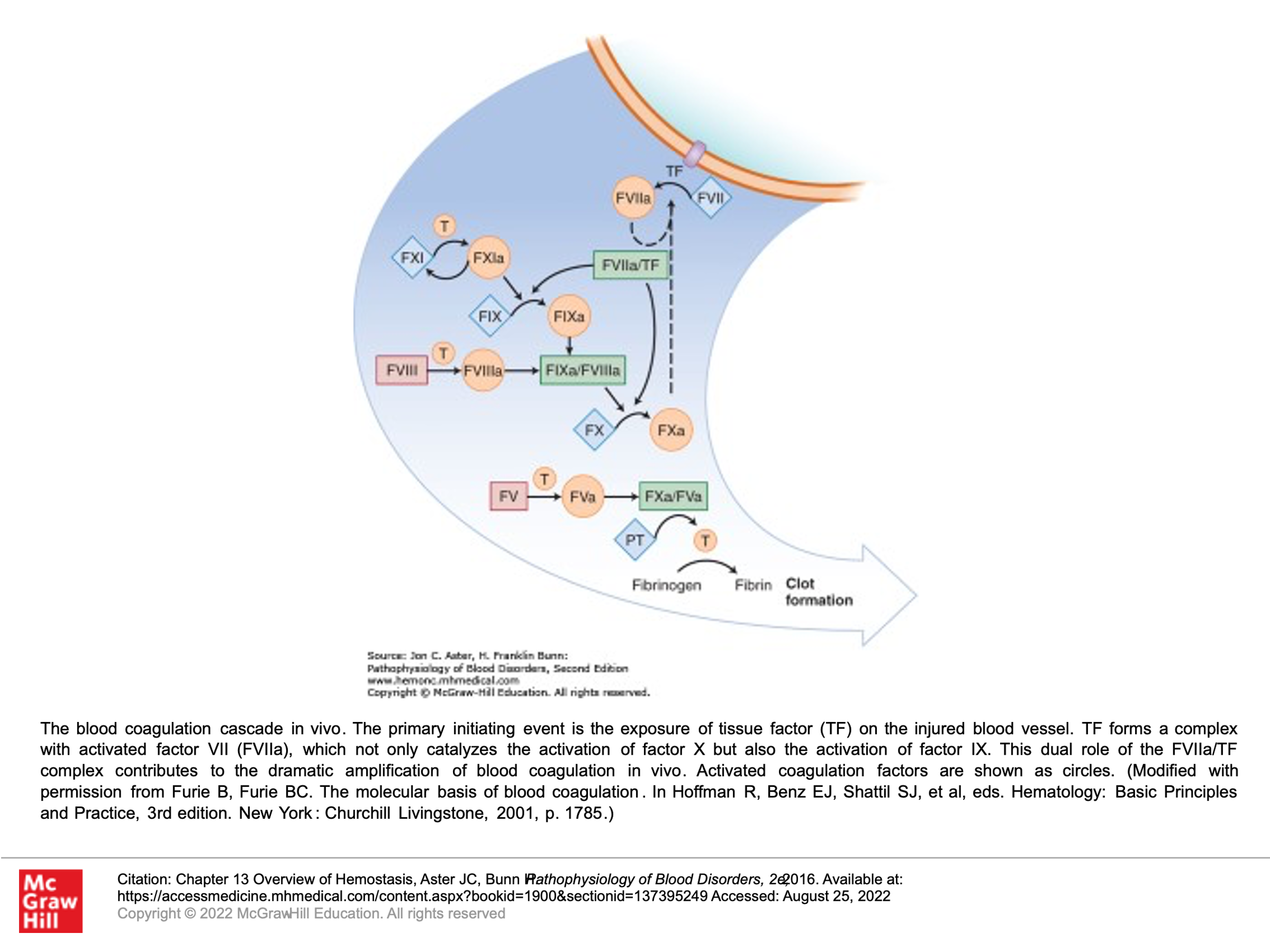
The blood coagulation cascade in vivo:
Note that in the actual human being, the clotting cascade is not quite as differentiated into 2 pathways, although the 2 pathways above explain how we understand results of the PTT and PT when we are measuring clotting. In vivo, the primary initiating event in the clotting cascade is exposure of tissue factor (TF) on the injured blood vessel (ie, the start of the extrinsic pathway above is thought to be the more common initiating event of clotting in vivo). TF forms a complex with activated factor VII, which not only catalyzes the activation of factor X, but also the activation of factor IX in vivo.
The clotting Cascade in Vivo:
Another depiction of In Vivo clotting concepts:
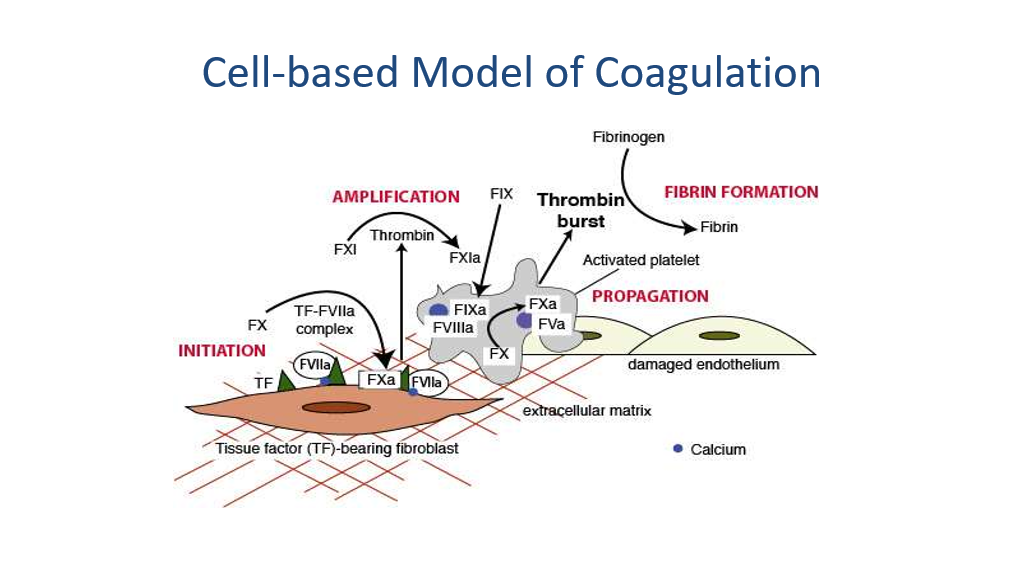
So, congrats! You have now mastered the clotting cascade.
But wait, if this keeps on going, then eventually one will clot off all their blood flow, which would be fatal!!! We have amplified clotting to stop bleeding, but now we need to shut it down so that we don’t keep clotting……
Good news. Built into the clotting cascade are ways to inhibit the cascade and shut it down.
Control of Clot formation: Inhibitors of the Clotting Cascade
There are several factors that inhibit the clotting cascade and are used to shut down the clotting cascade. They are as follows:
- Tissue Factor Pathway Inhibitor (TFPI): This protein is synthesized in endothelial cells and is present in plasma and platelets. It binds to Xa. Xa/TFPI complex then binds to VIIa/Tissue factor complexes and shuts down clotting via this process. As you can see, this mainly shuts down steps measured in the extrinsic pathway. This process starts after some Xa and some Thrombin have been generated. Hence, the clotting pathways measured by the intrinsic pathway still persist, despite actions of TFPI.
- Antithrombin (also known as Antithrombin III, also known as Heparin cofactor): Antithrombin is the most important of a number of plasma protease inhibitors in regulating hemostasis. Upon binding to endogenous glycoconjugates on the surface of endothelial cells or upon binding to the anticoagulant heparin, antithrombin undergoes a conformational change that significantly increases its activity as a serine protease inhibitor. It inactivates Thrombin, VIIa, IXa, Ix, and Xia as these activated factors are carried in the blood away from the developing thrombus.
- Protein C: Protein C, in the presence of its cofactor, protein S, is activated by thrombin that is bound to thrombomodulin on the surface of normal endothelial cells. Activated protein C (along with it’s cofactor, S) then inactivates Va and VIIIa.
- Protein S: As above, protein S is a cofactor necessary for protein C to function to inactivate Va and VIIIa.
(Small pearl to remember for later, when we talk about medications to treat clotting: Protein C, Protein S, Factor X, Factor IX, Factor VII, and factor II all require vitamin K-dependent carboxylation, and these factors share some other structural features. Vitamin K deficiency results in lower levels of Factors X, IX, VII, II, Protein C and Protein S.)
Note: Deficiencies of inhibitor proteins ATIII, Protein C and Protein S can result in disorders associated with an increased risk of thrombosis. I’m not aware of any clinical syndrome (probably because it wouldn’t be viable) of a deficiency in Tissue Factor Pathway inhibitor.
Putting the clotting cascade (intrinsic and extrinsic pathways) and the above inhibitors of the clotting cascade together!
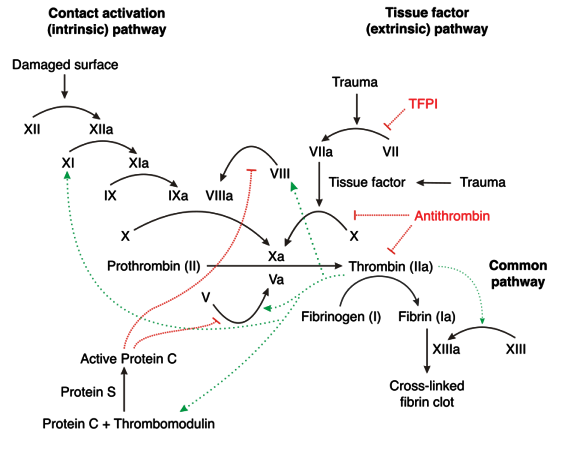
Clot Dissolution=Fibrinolysis
(Another control to restrict clotting)
The fibrinolytic pathway is triggered by clot formation: The fibrinolytic pathway promotes dissolution of unwanted clot in adjacent non-injured vessels as well as eventual removal of clot during and following healing of injured tissues. In this process, Tissue plasminogen activator (tPA) and urokinase-type plasminogen activator (urokinase), both of which are enzymes, convert plasminogen (another zymogen) into its active form, Plasmin. Plasmin then degrades Fibrin. Tissue plasminogen activator (tPA) is found on endothelial cells that line blood vessels, and its job is to prevent clot formation in intact blood vessels. Urokinase is an endogenous activator and is produced by kidney cells and can be recovered from urine. Fibrinolysis is the name of this process by which fibrin/clot is degraded.
There are 2 key things to note about Fibrinolysis:
- Plasmin cleaves fibrin into fragments. Plasmin is produced, as noted above, when tPA or urokinase (uPA) cleave thrombin-bound plasminogen to plasmin. Note that recombinant tPA and urokinase are drugs that are used to treat acute clotting events in certain situations such as Myocardial infarction, Stroke, and Pulmonary embolism. It is also important to note that Plasmin activity outside of the clot area is quickly inhibited by another protein: alpha2-antiplasmin.
- Thrombin is involved in keeping clots localized while fibrinolysis is occurring acutely elsewhere where clot is not needed. Thrombin, which of course is most present in the site of activated clotting, activates TAFI (thrombin-activatable fibrinolysis inhibitor). TAFI then protects the fibrin clot from lysis and stabilizes the blood clot.
NOTE: Overactivity of fibrinolysis can result in bleeding problems.
>>> Extra information, FYI: We can measure control of clot formation: The euglobin lysis time is a test that measures overall fibrinolysis. The euglobulin lysis time measures the time it takes for an in vitro clot to dissolve in the absence of plasmin inhibitors. Increased fibrinolytic activity is suggested by clot lysis that occurs in less than 2 hours. A shortened clot lysis time implies excessive fibrinolytic activity. Clinical bleeding is a possible consequence of excessive fibrinolysis. Excessive fibrinolysis may result from increased levels of tPA, increased plasmin activity, decreased levels of plasminogen activator inhibitor-1 (PAI-1) or decreased alpha2-antiplasmin activity. A shortened clot lysis time can also be seen with factor XIII deficiency, as fibrin won’t be crosslinked and will, hence, dissolve faster. A shortened clot lysis time will also be seen in the setting of hypofibinogenemia, as less fibrin clot will form in the first place when fibrinogen is lacking and thus less fibrin can form. <<<
Summarization of Hemostasis Processes
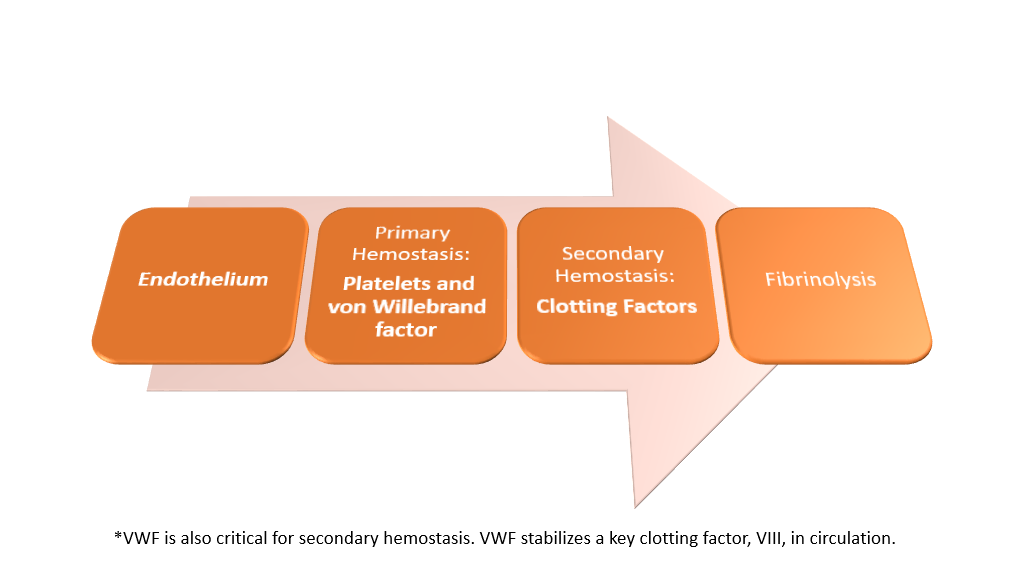
So, what can possibly go wrong???? Lots!!!!
We’ll now discuss disorders resulting in bleeding and clotting when we don’t want bleeding and clotting.
Disorders of Primary Hemostasis: Platelet disorders and Von Willebrand disease:
We will now review disorders or primary hemostasis.
Platelet disorders:
Optional textbook reading: Part II Hemostasis and Thrombosis, Chapter 14 (Platelet disorders)
As discussed above, vascular injury leads to thrombus formation. This begins with the formation of a platelet plug, which is then surrounded and strengthened by a fibrin polymer meshwork. Platelet adhesion to the exposed endothelium in a damaged blood vessel, etc., results in platelets binding to the area via interaction with Von Willebrand factor (VWF). This then results in 3 major reactions:
- Release of platelet granules
- Activation of platelet surface glyoprotein IIb/IIIa receptors
- Flip-flop of phospholipids from the inside of the platelet membrane to the outside, which supports coagulation.
Platelet Function will be affected by the following:
- Quantity of platelets. A low platelet count (aka thrombocytopenia) will increase risk of bleeding, as obviously there are less platelets circulating to plug holes in blood vessels. Clinically, we generally don’t see increased bleeding risks with low platelets until the platelet count drops below 50,000/ul.
- Quality of the platelets, which refers to how well platelets function. Even when platelets are present in normal numbers, if they don’t function properly then bleeding can occur. There are 2 main types of qualitative platelet function problems:
- Intrinsic platelet defects: These generally encompass inherited membrane or granule defects. This can include inherited receptor defects such as lack of ability to bind VW factor, etc.
- Extrinsic platelet defects: These generally encompass extrinsic factors that coat the platelets and/or impair platelet function. Uremia (seen in kidney failure, for example) is one cause of impaired platelet function. Certain paraproteins (antibodies) can cause this problem. Drugs such as aspirin or other NSAIDS impair platelet function.
Quantity of platelets: Low platelet number = Thrombocytopenia.
Recall that platelets are produced from megakaryocytes in the bone marrow. They are then released into the circulation. Platelets circulate, on average, about 8 to 9 days, and then senescent platelets are removed by the spleen.
Decreased production of platelets leads to low platelet numbers: This can be seen with bone marrow problems where megakaryocytes are affected such that they can’t produce platelets. There are several bone marrow problems than can cause this, which we won’t cover right here. Reduced production of thrombopoietin, the hormone that stimulates platelet production, also can result in decreased platelet production. Liver disease is a cause of decreased thrombopoietin production. Vitamin deficiencies (eg B12), toxins (eg alcohol), infections and marrow infiltration by other processes interfere with marrow production of platelets. Bone marrow testing in the setting of decreased platelet production can show decreased megakaryocytes in the bone marrow.
Increased destruction of platelets or consumption of platelets: This can be seen with autoimmune disorders where antibodies bind platelets, and then the spleen (doing its normal spleen job) takes the platelets out of circulation. Immune thrombocytopenia can be a primary platelet autoimmune disorder (AKA ITP or Immune thrombocytopenic purpura), where antibodies against platelets are made with a resultant isolated thrombocytopenia. Secondary Immune platelet destruction can also be seen with other autoimmune disorders (eg Lupus), lymphoproliferative disorders, some infections, and with use of certain drugs (example quinine, vancomycin, trimethoprim-sulfa). With massive bleeding, one can consume platelets faster then they are made. Platelets are activated/removed from circulation in the clotting seen with DIC (disseminated intravascular coagulation) and TTP (thrombotic thrombocytopenic purpura), which are disorders we’ll discuss further below.
In settings where platelets are consumed/taken out of circulation, bone marrow testing will typically show either normal numbers or increased numbers of megakaryocytes in the bone marrow.
Sequestration of platelets: This is also called splenic sequestration and occurs commonly in patients with significant splenomegaly. When the spleen enlarges (such as the case of liver disease with portal hypertension causing an enlarged spleen, for example), there is more of the spleen to remove senescent cells, and it does its job too well and removes more platelets than normal. (Bone marrow testing will often show normal megakaryocytes in this setting.)
Inherited platelet disorders that cause low platelets: These often can be differentiated by platelet size. Some of these problems also have inherent platelet function problems in addition to platelet number problems:
Macrothrombocytopenia (MYH9 Variants): Inherited disorders with low platelet numbers and large platelet size are rare. These include platelet glycoprotein abnormalities (eg Bernard-Soulier syndrome), deficiency of platelet alpha granules (eg gray platelet syndrome), May-Hegglin anomaly (involves the presence of abnormal neutrophil inclusions as well as platelet findings), and some kindreds with type 2B Von Willebrand disease (Montreal platelet syndrome). Some disorders are associated with glomerulonephritis, sensorineural deafness, or cataracts and a mutation of the gene encoding the heavy chain of non-muscle myosin-9 (MYH9-related disease).
Thrombocytopenia with absent radii (TAR): Inherited problem with normal-sized platelets, but low platelet numbers. Patients also have associated radial bone agenesis as well as other upper and lower limb defects. Affected infants usually require frequent platelet transfusions until approximately 1 year of age, by which time there is usually a spontaneous recovery of platelet production.
Congenital amegakaryocytic thrombocytopenia (CAMT): This presents in the newborn period with isolated severe thrombocytopenia and bleeding. Pancytopenia develops later in childhood.
Wiskott Aldrich Syndrome: This X-linked recessive disorder is characterized by immunodeficiency, severely dysfunctional platelets, and thrombocytopenia with small platelets (microthrombocytes). These platelets have a storage pool-like disorder with platelets that are unable to aggregate.
Uptodate.com has a nice summary of additional causes of thrombocytopenia in children, as there are other inherited disorders. Often, these are further broken down by disorders that cause small, normal, or large-sized platelets.
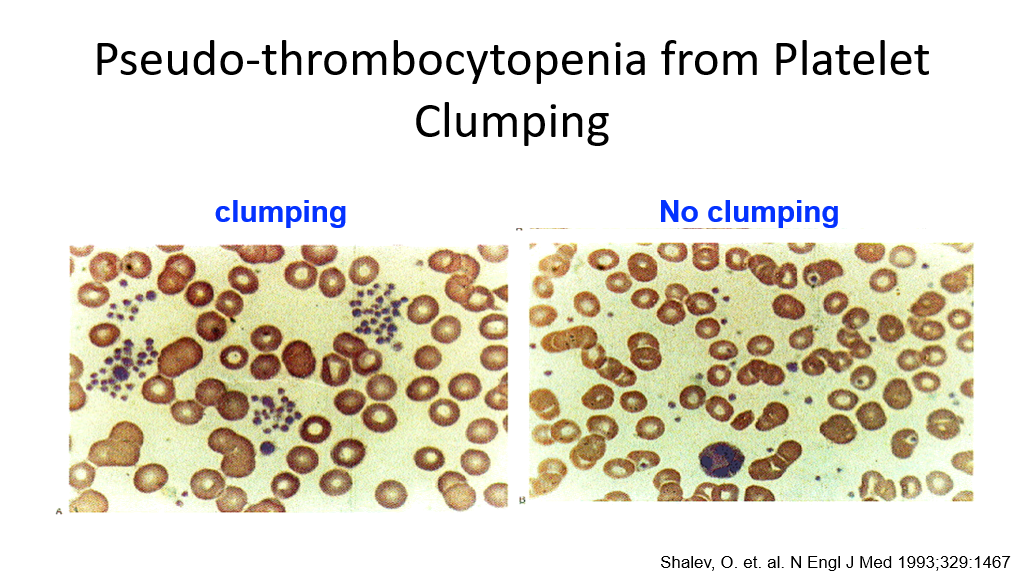
Note on Pseudothrombocytopenia: In addition to the above, there is a test tube issue that can result in a falsely low platelet count. When blood is drawn in a tube for blood counts, an anticoagulant such as EDTA is added to the tube. Certain anticoagulants can cause platelets to clump in some patients. Platelet clumps are then miscounted by automated machines as one large platelet, resulting in a miscount of platelets. This is not a true thrombocytopenia but can result in referrals to hematologists unnecessarily if a pathologist or doctor has not reviewed the blood smear, which will show platelet clumping in this case.
Immune Thrombocytopenic Purpura (ITP): An example of a quantitative platelet disorder
Immune Thrombocytopenic Purpura, also known as ITP, is an example of a quantitative platelet disorder which is not uncommonly encountered in clinical practice. This is an autoimmune disorder where patients make antibodies that target their own platelets. Platelets are then removed from circulation. The antibody is generally against certain platelet membrane glycoproteins. These antibody-coated platelets are then removed from circulation by the reticuloendothelial system (mainly the spleen). In children, ITP usually occurs as a post viral problem and resolves spontaneously. In adults, the problem may resolve spontaneously, but is more likely to become a chronic problem and may not be associated with a post viral phenomenon.
ITP, like other platelet consumption problems, results in increased platelet turnover, as platelets are taken out of circulation by the spleen when they are coated with antibodies.
ITP Treatment:
Given that ITP is an autoimmune problem, it makes sense that treatment involves immune suppression. We don’t’ generally treat patients with ITP unless their platelets drop below 30,000. The reason for this is that the risk of spontaneous bleeding due to low platelets doesn’t significantly increase until platelets drop below 20,000. In newly diagnosed patients with ITP who require treatment, the treatment of choice is steroids—generally prednisone, dexamethasone, or methylprednisolone. These steroids will suppress the immune system, and then the steroids can be weaned off. IVIg (Intravenous Immunoglobulin) can also be used in the acute setting. IVIG seems to work by fooling the spleen—overloading splenic FC receptors—such that the spleen can’t bind to antibody-coated platelets because it is overwhelmed by other foreign antibodies in circulation.
In patients that have relapsed/chronic ITP that requires treatment and are refractory to steroids (we don’t like to keep patients on long term steroids, as that is not healthy for them either), we use other forms of treatment including thrombopoietin receptor agonists such as romiplostim or eltrombopag. Recall that these drugs mimic thrombopoietin and stimulate increased platelet production and are used in ITP to offset the loss of platelets. Rituximab, a monoclonal antibody that lowers B-cells (recall that B-cells are the cells that mature to plasma cells and produce antibodies, including autoantibodies) can be used to lower antibody production. Other treatments for ITP include other immunosuppressants as well as splenectomy—surgery to remove the spleen and thereby stop splenic sequestration of antibody-bound platelets.
Qualitative Platelet Disorders:
Qualitative platelet disorders are disorders where platelets are present, usually in normal numbers, but don’t function like they are supposed to—which generally results in bleeding problems.
Qualitative Platelet disorders can be inherited (where a specific platelet process is inherently defective), due to systemic disease (eg kidney failure causing uremia), or drug-induced.
Drug-induced platelet dysfunction: This is by far the most common cause of platelet dysfunction in patients.
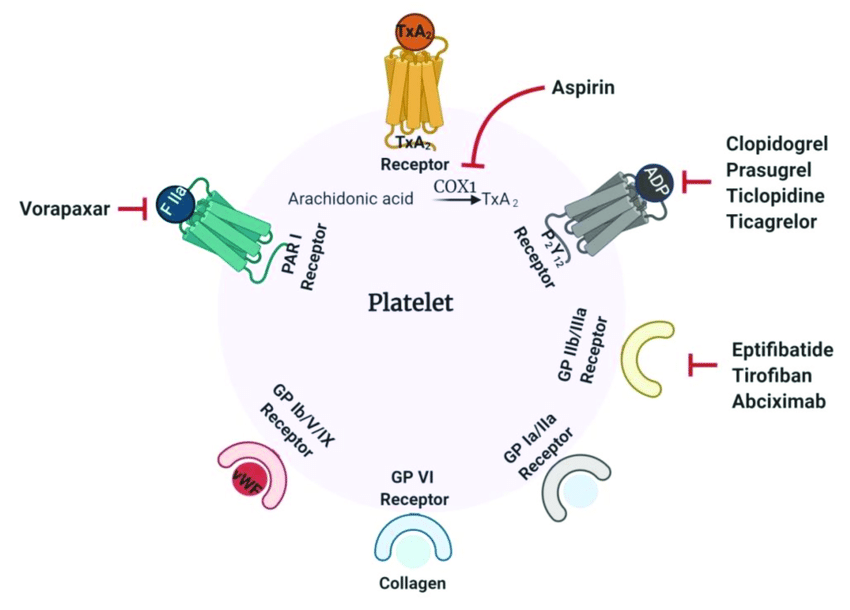
Drug-induced platelet dysfunction is particularly noted with the following drugs—which is why some of these drugs are used commonly to prevent clots in patients with cardiovascular disease:
- Aspirin: This is an irreversible cyclooxygenase inhibitor that inhibits platelet function. Note that because this is an irreversible inhibitor, the platelet is affected for its entire life once it has been exposed to this drug. Hence stopping aspirin won’t help platelets that are already inhibited by aspirin, and one must wait until new platelets are generated for the aspirin effects on platelets to be gone.
- Nonsteroidal Anti-inflammatory drugs (NSAIDs): These include ibuprofen, naproxen, and others. These are reversible cyclooxygenase inhibitors that will affect platelet function. Because they are reversible, their effects can wear off over time once they are stopped.
- ADP P2Y12 Receptor Inhibitors: These drugs include clopidogrel, prasugrel, ticagrelor, and function by inhibiting the ADP P2Y12 receptor on platelets such that platelets aren’t activated.
- GpIIb-IIIa receptor inhibitors: These drugs (eg. abciximab) inhibit platelet aggregation by preventing the binding of fibrinogen, von Willebrand factor, and other adhesive molecules to GPIIB/IIIa receptor sites on activated platelets. In addition to abciximab, tirofiban and eptifibatide inhibit GpIIb/IIIa receptors.
Inherited platelet disorders with Qualitative Platelet dysfunction:
- Disorders of platelet adhesion: Bernard Soulier syndrome (Platelets lack the Von Willebrand factor receptor, aka GpIb-IX-V receptor.
- Disorders of platelet aggregation: Glanzmann Thrombasthenia (Loss of the GpIIb-IIIa receptor).
- Disorders of platelet secretion: Generally these are disorders where one has platelet granule abnormalities, such as gray platelet syndrome, storage pool disease, etc.
- Signal transduction defects involving platelets.
- Other: See above re: inherited disorders with Quantitative platelet problems, as some of these also have Qualitative platelet problems.
Another disorder of Primary Hemostasis that is inherited: Von Willebrand Disease
Optional textbook reading: Part II Hemostasis and Thrombosis, Chapter 15 (Inherited Coagulation Disorders), which briefly reviews Hemophilia A, B and Von Willebrand Disease.
Recall that platelets adhere to exposed damaged endothelium in a blood vessel by binding to VW factor. The basis of primary hemostasis (platelet plugging) is due to this. In this setting, with vascular damage, subendothelium is exposed to blood. Von Willebrand Factor (VWF) binds to subendothelial collagen and then binds platelets via the platelet GpIb (again, note that the I is a roman number 1 in this case) receptor causing the platelets to adhere to the area of subendothelial damage.
We have previously discussed that problems with platelet plugging tend to result in mucocutaneous bleeding. A deficiency or lack of function of VWF also results in mucocutaneous bleeding/primary hemostasis problem.
Von Willebrand disease is a family of disorders involving deficiency or lack of function of VWF (von Willebrand factor). It is the most common inherited bleeding disorder and is inherited in an autosomal dominant pattern. Males and females are affected equally. Clinical manifestations of VW disease include mucosal bleeding (epistaxis, gum bleeding, heavy menstrual bleeding, post-partum hemorrhage, bleeding after dental extractions, bleeding after tonsillectomy, and GI bleeding), excessive bruising, hemorrhagic ovarian cysts, and perioperative bleeding. Note that VWF is also a carrier/stabilizer protein for Factor VIII. Hence, with severe VWF deficiency, Factor VIII levels will also be low. Because of this, in severe VW disease, one can also have hemarthroses (like hemophilia A) due to low Factor VIII. Also note that Von Willebrand Factor is present in the blood in different size multimers.
Acquired Von Willebrand Syndrome:
In addition to the inherited form, VWD can be acquired.
Acquired deficiency and/or dysfunction of VWF can be seen due to the following:
- Autoantibodies against VWF
- Some medications (example: Valproic acid).
- Severe hypothyroidism (this is exceedingly rare).
- Shear Stress Destruction of Von Willebrand Factor Multimers. This can be seen in aortic stenosis and is a common cause of bleeding in that disorder. (Heyde’s syndrome involves Aortic stenosis, acquired Von Willebrand syndrome, and GI bleeding from angiodysplasias in the gut.) Shear stress destruction of VWF multimers can also seen when one has a perivalvular leak after a heart valve repair. It can be seen with some ventral septal defects. It is also seen with left ventricular assist devices and extracorporeal devices such as ECMO.
Review: Bleeding Pattern seen in disorders of primary hemostasis (platelet or Von Willebrand factor disorders):
Note that bleeding seen in patients with platelet disorders/disorders of primary hemostasis (this includes bleeding with most types of Von Willebrand disease due to lack of/malfunctioning VWF) is different than seen in patients with clotting cascade problems. Bleeding with platelet disorders/primary hemostasis disorders generally is that of mucocutaneous bleeding such as bruising, ecchymosis, epistaxis, gum or other oral bleeding, heavy menstrual bleeding, and GI/GU bleeding.
Bleeding problems due to the problems with the clotting cascade (problems with secondary hemostasis):
In addition to problems with platelet function, as discussed above, bleeding disorders can also be due to problems with clotting cascade itself (in other words, problems with secondary hemostasis). These are coagulation factor disorders. These can be acquired or inherited.
Acquired problems resulting in abnormal secondary hemostasis:
- Vitamin K deficiency: This results in decreased production of vitamin K-dependent clotting factors X, IX, VII, and II (prothrombin). Note that protein C and protein S (which are involved in suppressing the clotting cascade) are also vitamin K dependent. However, with significant vitamin K deficiency, bleeding is more problematic, due to multiple clotting factors being deficient in the clotting cascade. Vitamin K deficiency can be due to malnutrition or due to malabsorption. Warfarin is a drug that interferes with production of vitamin K-dependent clotting factors (as well as protein C and S). Neonates have low vitamin K stores at birth. Because of this, they are generally given vitamin K (shot given IM) at birth to reduce risks for bleeding, particularly the most serious risk of spontaneous intracranial bleeding due to low vitamin K-dependent clotting factors. Vitamin K can be given either orally, Intramuscularly (IM), or IV. In neonates, it is given IM for quick and adequate absorption.
- Liver disease: All clotting factors except for VIII are made in the liver. Hence clotting factors (other than VIII) will be low with significant liver disease. (As an aside, VIII and Von Willebrand factor are made by endothelial cells.)
Inherited Clotting Factor Disorders resulting in abnormal secondary hemostasis:
Optional textbook reading: Part II Hemostasis and Thrombosis, Chapter 15 (Inherited Coagulation Disorders), which briefly reviews Hemophilia A, B and Von Willebrand Disease.
These are the hemophilias and there are two types of hemophilia that you need to know about (other hemophilias are extremely rare). Both are X-linked in inheritance.
- Hemophilia A: this is an X-linked bleeding disorder due to deficiency of factor VIII.
- Hemophilia B: This is an X-linked bleeding disorder due to deficiency of factor IX.
Hemophilia A and B are seen world-wide and in all racial groups. This because the spontaneous mutation rate is high (30% of cases are due to a new mutation, which is then subsequently inherited).
Question: Is the PTT or PT prolonged in Hemophilia A or in hemophilia B?
Answer: PTT is prolonged, but not PT. (You should be able to answer this by reviewing your clotting cascade above.)
Hemophilia
- Hemophilia A (factor VIII deficiency)
- Hemophilia B (factor IX deficiency)
- Phenotypically identical:
- Hemarthroses
- Hematomas
- Gastrointestinal and genitourinary leeding
- Severe: <1% factor
- Moderate: 1%-4% factor
- Mild: >4% factor
- Screening Assays: ↑ partial thromboplastin time, normal prothrombin time
In a patient with XY chromosomes who presents with a bleeding tendency that fits with a secondary hemostasis problem and who is then found to have a normal PT and abnormal PTT, the next diagnostic step would be to do specific factor VIII and factor IX lab assays, as this type of history would point to a hemophilia diagnosis. The factors are measured by their activity in clotting assays in the lab. Basically, Factor VIII- or Factor IX-depleted plasma is used as a substrate and the clotting time with the patient plasma is compared to the clotting time of normal pooled plasma. Normal factor VIII activity is about 50-150%. Normal Factor IX activity is about 50-200%.
Note, due to X inactivation in patients with XX chromosomes, XX chromosome patients can have variable risk for bleeding if they have inherited one of the mutant X chromosomes. Many carriers of hemophilia (XX individuals with one mutated X-chromosome for hemophilia) will have lower Factor VIII or Factor IX activity levels. However, a normal factor level does not exclude the carrier state. Genetic testing is needed to diagnose a carrier state in XX individuals.
Recall that with platelet problems, mucocutaneous bleeding is more common. This is not the case with hemophilia. In hemophilia, the more common sites of bleeding are soft tissue, joints, and muscles. Life-threatening bleeding can be seen with bleeding in the CNS, neck/throat (obstruction of airway), GI bleeding, and retroperitoneal bleeding. Hemophilia presentation typically involves recurrent bleeding eventually leading to the diagnosis. The onset of bleeding/severity of bleeding will depend upon the factor level in hemophilia A and B. (Yes, nothing is simple in medicine. Patients with hemophilia A or B can have variable bleeding, as there are variants with more or less severe factor VIII or IX deficiencies.)
- Severe bleeding risk is generally seen when the Factor VIII or IX activity level is < 1%. (Severe Hemophilia A or B).
- Moderate bleeding risk is generally seen when the Factor VIII or IX activity level is 1-5%. (Moderate Hemophilia A or B).
- Mild bleeding risk is generally seen when the Factor VIII or IX activity level is 6-40%. (Mild Hemophilia A or B).
Note, some patients with mild hemophilia A or B don’t actually have significant bleeding and may be diagnosed later in life or not at all!
Treatment of Hemophilia A or B:
Treatment involves replacement of the factor that is low. In the past, patients were treated at time of bleeding. However, we now know that it is better to prevent bleeding in order to prevent long term complications of joint bleeds. Hence, state of the art is to do regular clotting factor infusions to prevent bleeding (aka prophylaxis). Because of this practice of prophylaxis with factor infusions, patients with hemophilia can now reach adulthood without joint abnormalities. Further advances in the therapy of hemophilia include the development of longer-acting factor replacement products (which don’t have to be given as frequently), alternative hemostatic agents that can be given subcutaneously, and gene therapy.
Risks to treatment: In the old days, factor replacement involved giving pooled factor products culled from plasma from many blood donors, and patients were at risks for infections from HIV, hepatitis, etc. Now days, we have recombinant products/processed products such that risk of infections are minimal to none. However, Patients who are given clotting factors that they don’t have are at risk of developing antibodies to the administered clotting factor, as such factors can be seen as foreign to their immune systems. These antibodies basically cause the factor to be destroyed and are called clotting factor inhibitors. (Such inhibitors can also be acquired in other individuals in other settings as well, leading to an acquired hemophila due to inhibitors.)
Clinical Scenarios with Low platelets but associated with Clotting: These are BAD, and you don’t want to miss these in clinical practice as the consequences can be life-threatening….
Above, we covered disorders of primary and secondary hemostasis in the form of platelet disorders, Von Willebrand disease, and two types of hereditary hemophilia’s. There are a couple more disorders we’ll highlight now. Disseminated Intravascular Coagulopathy (DIC) involves simultaneous bleeding and clotting problems. Thrombotic Microangiopathies are disorders that result in low platelets and clotting. These problems are BAD, so you don’t want to miss them in clinical practice!
Disseminated Intravascular Coagulation (DIC):
Optional textbook reading: Part II Hemostasis and Thrombosis, Chapter 16 (Acquired Coagulation Disorders, section on DIC)
DIC, as its name describes, involves clotting occurring intravascularly throughout the body. This is a medical emergency and can result in bleeding or clotting (or both) in a patient and is life-threatening. It is thought that this occurs when blood is exposed to tissue factor (TF) or TF-like substances. This can occur in the setting of trauma, obstetric accidents/OB complications, diffuse vascular injury, increased endothelial permeability, activated monocytes, and via tumor cells—they may have their own protein that activates coagulation. DIC is a pathologic response to an injury which involves activation of blood clotting and fibrinolysis which are so robust that they are not controlled by the normal physiologic control mechanisms. With DIC we see abnormal clotting. Due to consumption of clotting factors and platelets activated by clotting, we can also see bleeding in patients with DIC, in addition to clotting. Laboratory findings in patients with DIC include the following:
- Elevated PT
- Elevated PTT
- Decreased platelet count
- Decrease in individual clotting factors
- Presence of fibrin degradation products ( D-dimers, FDPs).
- Presence of Schistocytes (fragmented RBCs) due to shearing of red cells on the intravascular clots.
Treatment of DIC generally involves treating the underlying cause and is otherwise supportive. If patients are bleeding, then factor support (platelets, plasma, etc.) is given. If patients are clotting, anticoagulation can be done, generally using heparin.
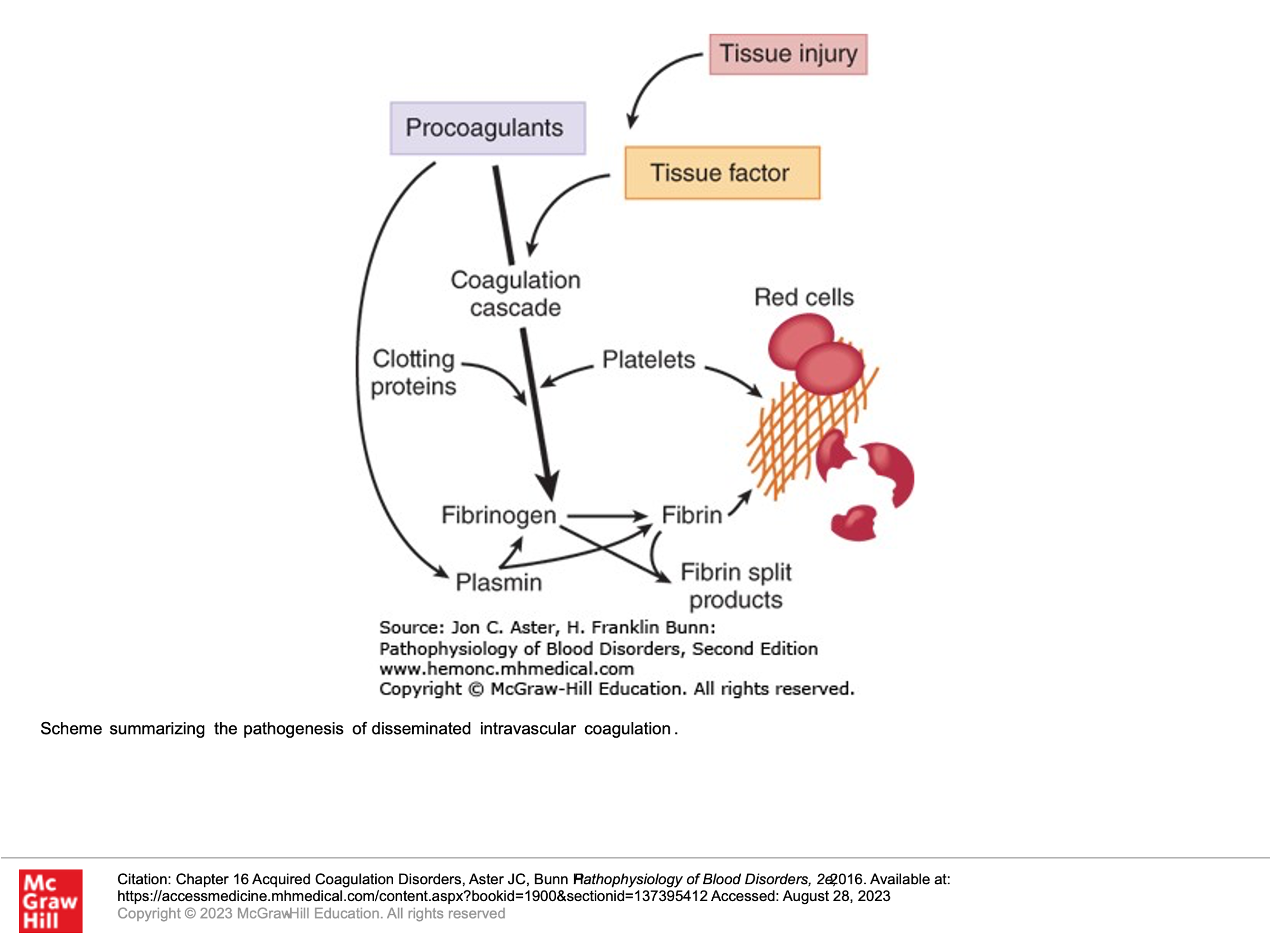

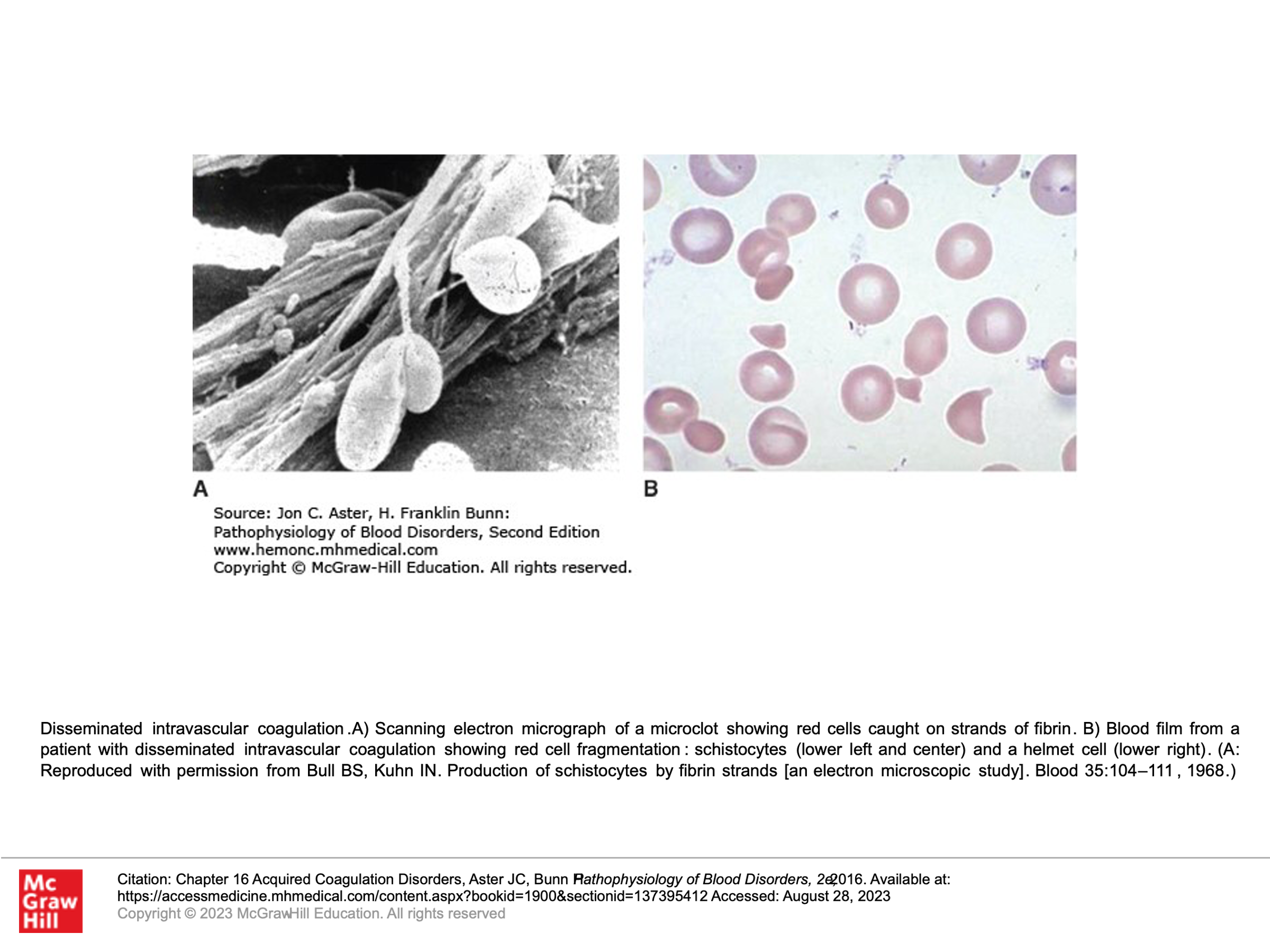
TABLE 16-1 Major Disorders That Trigger Disseminated Intravascular Coagulation
|
Vascular Damage Leading to Release of Tissue Factor |
|
Bacterial sepsis Gram-negative organisms Meningococcus Pneumococcus Gram-positive organisms Metabolic stress Acidosis Shock Heat stroke |
|
Release of Tissue Factor from Injured or Pathologic Tissue |
|
Obstetrical complications Placental abruption Retained placenta/fetus Placenta previa Amniotic fluid embolus Preeclampsia/eclampsia Malignancies Solid tumors, mucin-secreting adenocarcinomas Acute promyelocytic leukemia Severe burns |
Thrombotic Microangiopathies (TMA), also called Microangiopathic Hemolytic Anemia (MAHA):
Thrombotic Microangiopathies are clinical syndromes defined by low platelets, fragmentation of red blood cells (schistocytes), and organ damage due to the formation of microscopic blood clots in capillaries and small arteries. There are 2 main types of Thrombotic Microangiopathies:
- Thrombotic Thrombocytopenic Purpura (TTP)
- Hemolytic Uremic Syndrome (Typical HUS)
- Hemolytic Uremic Syndrome is then further categorized as follows:
-
Typical HUS
-
Atypical HUS
-
Secondary HUS
-
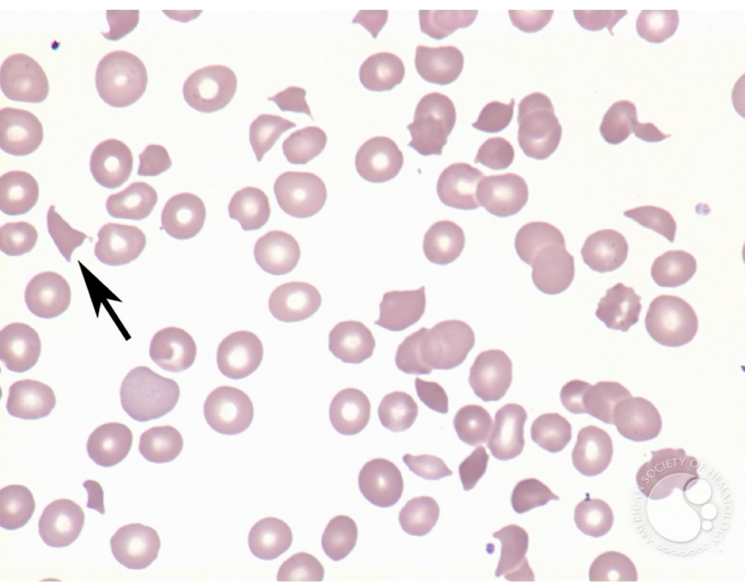
Peripheral Blood Smear of a patient with a Microangiopathic Hemolytic Anemia (MAHA)
Remember MAHA = microangiopathic hemolytic anemia (fragmentation hemolysis, shistocytes on smear):
Thrombotic Thrombocytopenic Purpura (TTP)
To understand TTP, one must first understand that Von Willebrand Factor (VWF) is present in blood in large multimers or polymers of VWF. As these polymers circulate, shear stress causes them to unfold in the peripheral blood. These partially unfolded multimers then expose cleavage sites in the multimers. These cleavage sites are then cleaved by VWF proteinase (also called ADAMTS13 enzyme or ADAMTS13), which is found in normal circulating blood. This results in various lengths of VWF in the blood. In TTP, there is a loss of ADAMTS13, generally due to antibodies to ADAMTS13 that develop. In this setting, partially unfolded VWF multimers have exposed cleavage sites, but cannot be cleaved. These unfolded circulating aggregates of VWF then bind to platelets and lead to platelet aggregates in the circulating blood that plug capillaries and small vessels. The following slides illustrate this:
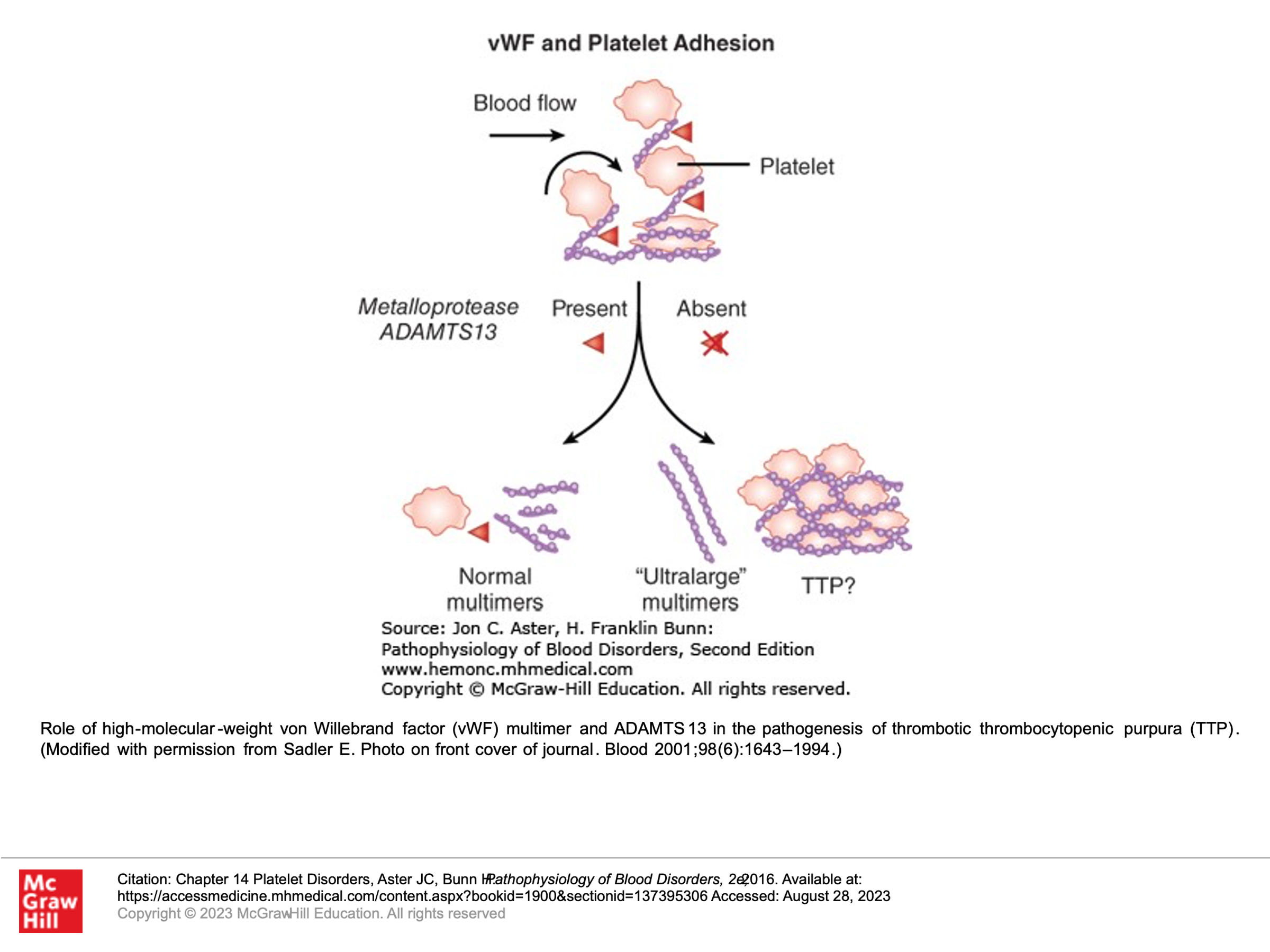
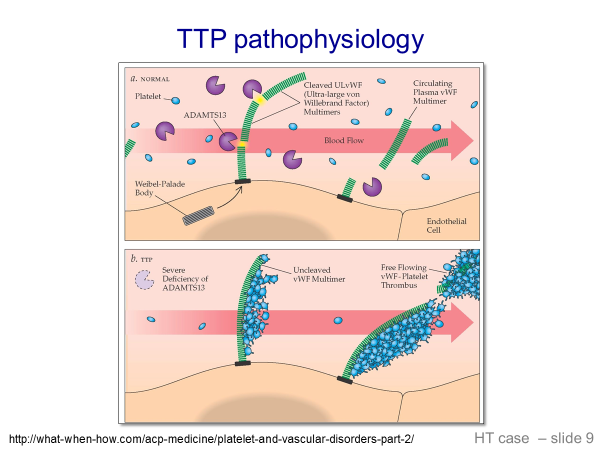
Clinical and Laboratory findings in TTP
Clinical manifestations: Mental status changes, fatigue, ecchymoses, and fever. Note that renal failure/other organ failure findings are less common with TTP (as compared to HUS).
Laboratory findings: Thrombocytopenia, microangiopathic hemolytic anemia—schistocytes with elevated LDH and bilirubin, normal PT and normal PTT, elevated creatinine may be seen, decreased ADAMTS13. Note that ADAMTS deficiency is considered evidence of an inhibitor or antibody to ADAMTS13 unless a congenital deficiency of ADAMTS13 is known in the patient.

Treatment of TTP:
Therapeutic Plasma Exchange: The hallmark of treatment is Therapeutic Plasma Exchange (TPE). This involves a dialysis-like procedure where the patient’s plasma is removed and is replaced with donated plasma. In removing the patient’s plasma and replacing this with normal plasma, large VWF multimers are removed, antibody to ADAMTS13 is removed (if it is present) and functional ADAMTS13 is provided in the normal plasma.
Caplacizumab: This is an anti-VWF antibody that is given by subcutaneous injection. It argets the A1-domain of VWF, inhibiting the interaction between VWF and platelets. This, in turn, reduces both VWF-mediated platelet adhesion and platelet consumption. One can easily deduce, thus, that the main risk of this medication is bleeding due to decreased interaction of platelets with VWF. Caplacizumab is approved for treatment of TTP in conjunction with plasma exchange.
Immune Modulation: This has a secondary role in treatment of TTP and generally is used in recurrent TTP in the setting of autoimmune destruction of ADAMTS13. For example, Rituxmab therapy to deplete B-cells can lower antibody production of the Anti-ADAMTS13 antibody.
Hemolytic Uremic Syndrome (HUS):
HUS is a syndrome where we see microangiopathic hemolytic anemia and thrombocytopenia and small vessel thrombi (like we see in TTP above), but no evidence of low ADAMTS13. This syndrome more commonly affects the kidneys. Mental status changes are much less common in HUS than in TTP, as is fever. It has 3 different subcategories:
- Typical HUS: This is usually due to shiga toxin-producing E.coli HUS (0157:H7). When you hear of E.coli outbreaks causing illness, one of the problems due to this E.coli causing infection is “typical HUS”. This is classically a disease of children. Patients with E.coli infections from food usually present with watery diarrhea that progresses to bloody diarrhea, along with severe crampy abdominal pain and nausea/vomiting. These symptoms are due to the E. coli infection in the gut. Following resolution of GI symptoms (usually 2-14 days after the onset of diarrhea), patient who go on to have typical HUS due to the E.coli then develop thrombocytopenia and acute kidney injury. Patients at this point present often with a “rash” that is not really a rash, but instead is purpura, and hematuria. Management of Typical HUS is supportive care. About 25% of patients develop chronic kidney disease, HTN, and proteinuria. Of note, antibiotic therapy is controversial, as rapid killing of the E.coli is thought to induce more release of shiga-toxin and more risk for HUS. Antibiotics are not used unless there are other clear indications of ongoing infection that warrant antibiotic treatment other than HUS.

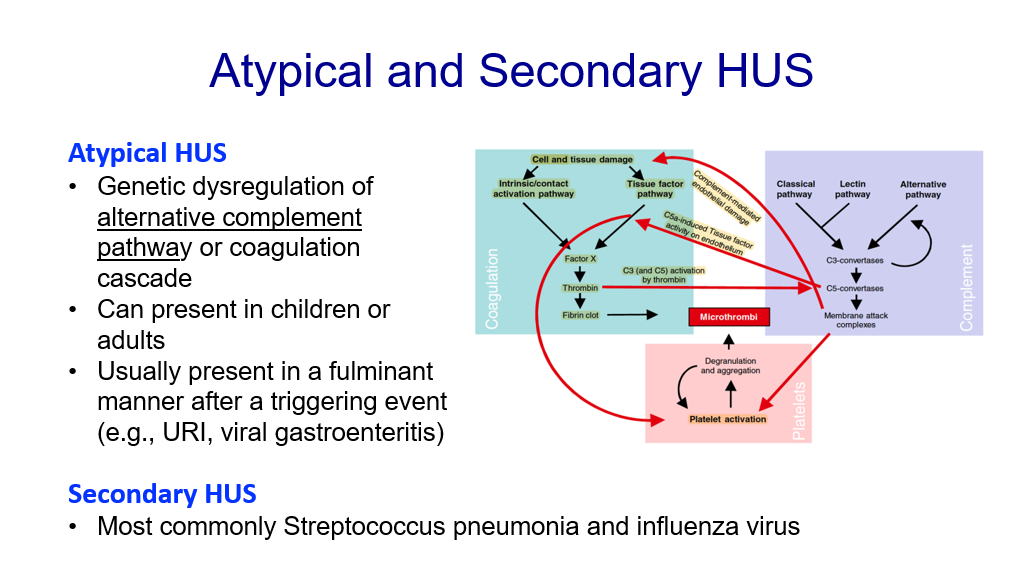
- Atypical HUS: This is due to genetic dysregulation of the alternative complement pathway (which you covered in infection and immunology). It usually presents in a fulminating manner after a triggering event such as an upper respiratory infection or viral gastroenteritis. Treatment of complement-mediated HUS utilizes the drug Eculizumab, which is a monoclonal antibody that targets the Complement 5 protein to inhibit complement. Another form of atypical HUS can involve the coagulation cascade.
- Secondary HUS: This is HUS due to another cause, most commonly streptococcal pneumonia and influenza virus.
Summary of Thrombotic Microangiopathies
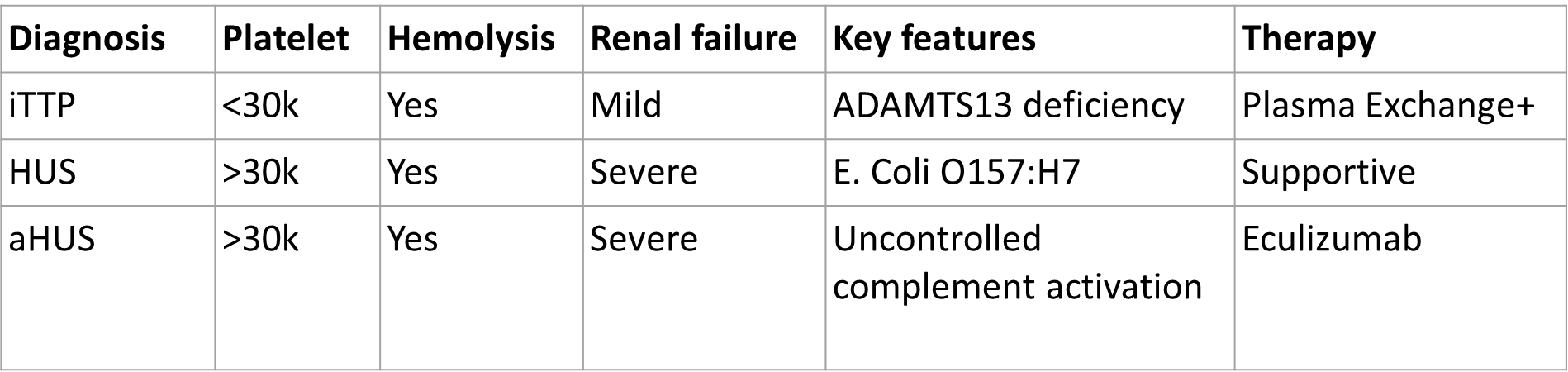
It can be challenging to diagnose and treat the various Thrombotic Microangiopathies (TTP, Typical HUS, Atypical HUS, Secondary HUS). The main challenge is deciding whether the patient has TTP, typical HUS, or atypical HUS. Does the patient need urgent therapeutic plasma exchange? Should the patient receive anti-complement therapy? Are there other treatment options to consider? Patients with a high index of suspicion for having TTP are given therapeutic plasma exchange up front. Patient with a high suspicion for having complement-mediated TMA and low compliment levels are treated with anti-complement therapy. Patients with a high suspicion for HUS due to shiga-toxin E.coli are treated supportively. Patients with other systemic disorders thought to cause HUS are treated for those disorders. It can be very challenging in the acute clinical setting to decide therapy for patients with Thrombotic Microangiopathies initially! Students should understand the typical clinical presentations, underlying pathogenesis, and laboratory features of these disorders. Here is a table reviewing some key features of each.
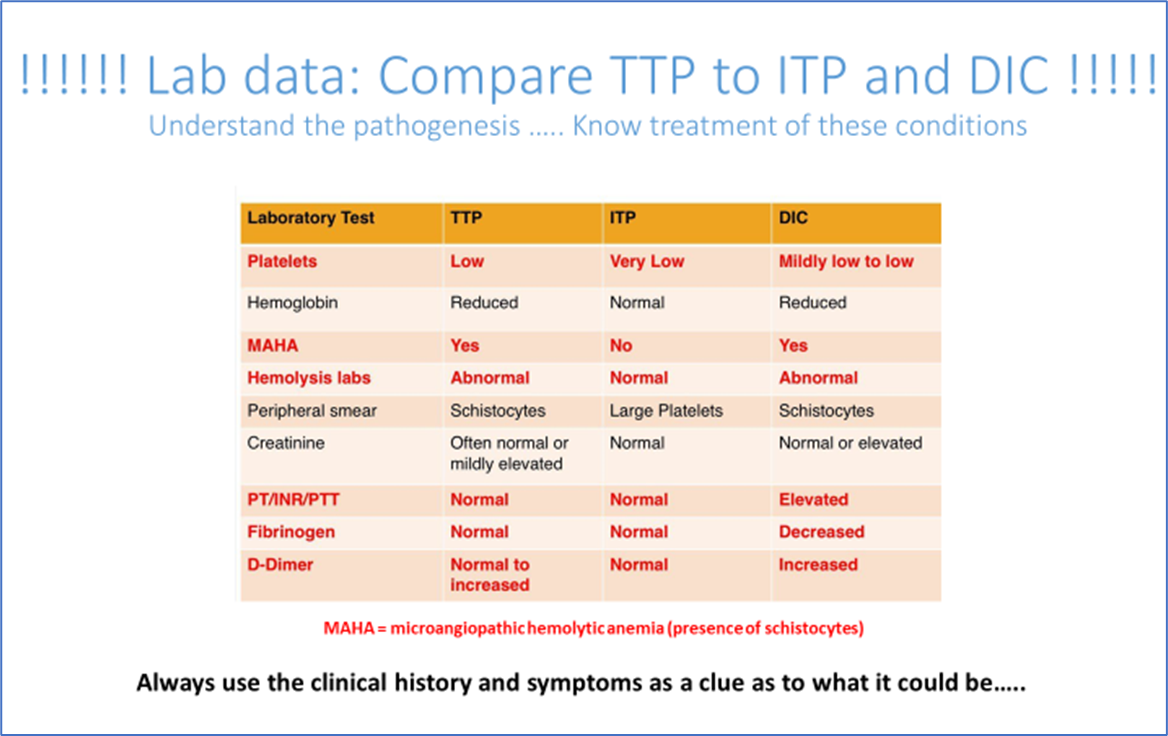
In addition, here is a table that reviews some laboratory differences between TTP, ITP, and DIC:
Authors and Contributors:
Corliss Newman, MD authored this course pack material based partly upon slide lecture material prepared by Rebecca Kruse-Jarres, MD/MPH. Nicholas Burwick, MD and Kristi Rice, MD provided editing assistance.