
3 Approach to Anemia
Learning Objectives
- Develop a coherent approach to the clinical and laboratory evaluation of patients with anemia based on classifying anemias as due to underproduction of red blood cells or excessive destruction of red blood cells (under production anemia vs excessive destruction anemia).
- Define the common causes of microcytic, normocytic, and macrocytic underproduction anemias.
- Explain systemic iron homeostasis and the pathophysiologies of hemochromatosis, iron deficiency, and anemia of chronic inflammation.
USMLE Content Descriptors: Cell/tissue structure and function; anemia, cytopenias, and polycythemia anemias; decreased production; hemolysis; other causes of anemia, hormones produced by or acting on the kidney, vitamin deficiencies and/or toxicities.
Review of Erythropoiesis, Definition of Reticulocytes, and Heme Synthesis
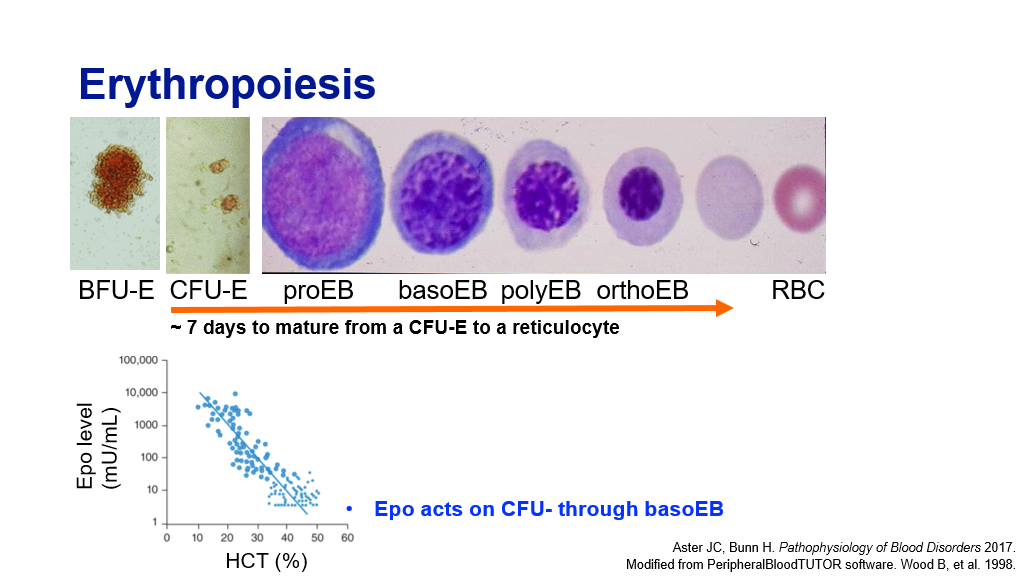
Erythropoiesis:
We have previously discussed how Red blood cells are produced from precursor cells in the bone marrow, in a process called erythropoiesis. Recall that EPO acts on CFU-E thru basophilic erythroblast stage. Basically, the kidney senses an oxygen need and produces EPO. This in turn acts on erythroid progenitor cells to increase RBC production. It takes about 7 days for a CFU-E to mature to a reticulocyte. Hence, it takes about 7 days to mount a reticulocytosis in response to an oxygen need.
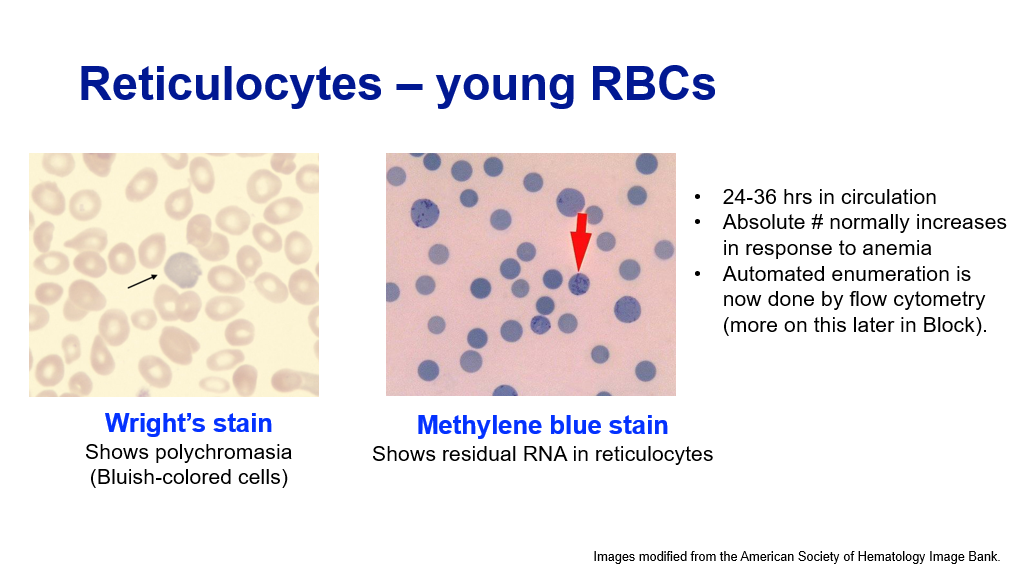
Reticulocytes:
Reticulocytes are red blood cells that are the earliest anucleate cells. (Recall this is the stage of RBC maturation after the nucleus from the orthochromic erythroblast has been extruded.) Reticulocytes still contain some bits of mRNA in their cytoplasm. These cells also require a few passes thru the spleen before they finally have a biconcave shape and become mature red blood cells. They can be detected in circulation as somewhat bluish colored RBCs on a Wright’s stain peripheral blood smear. Staining of RBCs with methylene blue stain will show residual RNA in reticulocytes.
Review of Heme Synthesis:
As RBCs mature, hemoglobin is being produced in the RBC cytoplasm. This involves production of the globin genes and the heme moiety. Recall that heme moiety synthesis involves eight enzymes, four of which are cytoplasmic and 4 of which are mitochondrial. The initial step takes place in the mitochondria where succinyl-CoA and glycine, in the presence of the enzyme 5-aiminlevulinic acid synthase-2 (ALA-S2) yields the product delta-aminolevulinic acid (ALA). This first step is the rate limiting step in the production of the heme moiety and is regulated by iron availability. ALA then migrates from the mitochondria to the cytoplasm, where the next 4 steps in heme synthesis occur, resulting int the production of coproporphyrinogen III. This product then migrates back to mitochondria, where the last 3 steps leading to production of heme occur. The last enzyme in the pathway, ferrochelatase, inserts iron into protoporphyrin IX to produce the final product, Heme (the heme moiety). This last step is the next rate limiting step in the production of heme. Note that in states of iron deficiency, ferrochelatase will insert zinc into the protoporphyrin ring. Students should know the first and last steps in the Heme production pathway.
Summary of a Normal RBC Life Cycle/Lifespan:
- Production/maturation: Occurs in the bone marrow. Maturation from a progenitor to an enucleate RBC takes about 14 to 21 days. Daily RBC production by the bone marrow is about 200 billion RBCs a day.
- Circulation in peripheral blood: Once the RBC is at the reticulocyte stage, it is released into peripheral blood. In 1 to 1.5 days after it is released into the blood, a reticulocyte matures into a mature RBC. A mature RBC lives about 90 to 120 days in circulation.
- Senescence: When a red blood cell reaches the end of its life span and becomes fragile, etc., it is taken out of circulation by the spleen. Its components are metabolized/recycled, provided for production of new RBCs, etc.
Anemia
What is anemia? Answer: Not enough red blood cells (low normal red blood cell measurements).
Red blood cells are measured in blood in a variety of ways:
- Red blood cell count: Number of red blood cells
- Hematocrit: Percent of whole blood that is red cells (Normal is about 42%).
- Hemoglobin: Measurement of hemoglobin in blood. (Normal is about 12-13 g/dL.) The World Health Organization defines anemia using hemoglobin, and considers a hemoglobin of < 13 in males or < 12 in females to indicate anemia.
Note, when a physician orders a CBC lab tests (complete blood count), certain results of that test are measured, and other certain results are calculated. It is important to understand this as we interpret the test results:
Measured Parameters of the CBC (complete blood count):
- Platelets
- Red blood cells
- White blood cells and differential (different types of white blood cells)
- Hemoglobin (HGB)
- Mean corpuscular volume (MCV)—this is the mean size of red blood cells
- Reticulocyte count
Calculated Parameters of the CBC:
- Hematocrit (HCT): RBC x MCV/10
- Mean corpuscular hemoglobin (MCH): this is the mean of the hgb contained in each RBC, and is calculated: HGB x 10/RBC
- Mean Corpuscular Hemoglobin Concentration (MCHC): HGB x 100/HCT
- RBC distribution width (RDW): This tells us how much the cells vary in size around the mean.
Anemia can be due to either not making red blood cells well, due to destruction of red blood cells, or simply due to blood loss. We tend to classify anemia as two basic types of anemia:
- Underproduction anemia: Lack of production of red blood cells. This generally results in a low corrected reticulocyte count because the bone marrow isn’t making new red blood cells like one would expect in the setting of anemia. Lack of production can be due to a lack of nutrients such as iron, B12, or folate. Underproduction anemia can also be due to a bone marrow problem where the bone marrow isn’t making cells as well. The (corrected) reticulocyte count is low in this setting.
- Increased production anemia: The body is trying to make new blood cells but can’t keep up with blood loss or destruction. Anemia is due to increased red cell destruction (such as hemolysis) or due blood loss with adequate building blocks to make RBCs. In this case, the bone marrow has the nutrients and the ability to make the cells, but blood loss is exceeding this ability. In this setting, the corrected reticulocyte count is high, as the bone marrow revs up production of RBCs, and hence more reticulocytes are made. The (corrected) reticulocyte count is high in this setting.
Knowing all of this, we can simplify how we approach the process of evaluating a patient with Anemia by the following steps:
- What is the clinical history? (Is the patient experiencing blood loss, have they been ill, etc?)
- Proceed to the laboratory assessment.
The Laboratory Assessment of Anemia has five steps, and by doing these steps, one can generally solve the mystery about why a patient is anemic. Here are the 5 steps:
- Are the RBCs produced appropriate for the degree of anemia? (In other words, is the bone marrow producing reticulocytes appropriately? To answer this, one needs to know what the Corrected Reticulocyte Count is. We’ll discuss what we mean by “corrected” reticulocyte count in a minute.
- If the corrected reticulocyte count is low (IE, the bone marrow is not making retics like we would expect it to in the setting of anemia), are the RBCs maturing appropriately? (Hint: We can get a clue about this by the size of the Red blood cells, so we want to know what the MCV is in this setting.)
- If the corrected reticulocyte count is high, then do the labs support hemolysis?
- Are any other cell lines (WBCs, platelets) involved?
- Look at the peripheral blood smear.
To understand these 5 steps above, one first needs to understand the concept of a “corrected” reticulocyte count. As you recall reticulocytes are the newly minted RBCs that have just been released from bone marrow and still have a little bit of mRNA in their cytoplasm. These cells can be measured and are generally measured as a percentage of total RBCs. In the setting of anemia, one would expect the percentage of retics to rise relative to the other RBCs, if the bone marrow is appropriately revving up production of red blood cells. So, basically, the corrected retic count tells you if the bone marrow is appropriately producing RBCs in response to the anemia. But, of course, whether your total RBC count is normal or low, the total percentage of RBCs in a given patient is still 100% for that patient. Hence, whether you are anemic or not, your RBC count is the 100% against which the percentage of retics is compared. Because of this, one must correct the reticulocyte percentage to account for the degree of anemia.
The graphic below shows this nicely, with the caveat that the math is a bit off in that they are saying 1/3 = 30% (instead of 33%) to simplify the math.
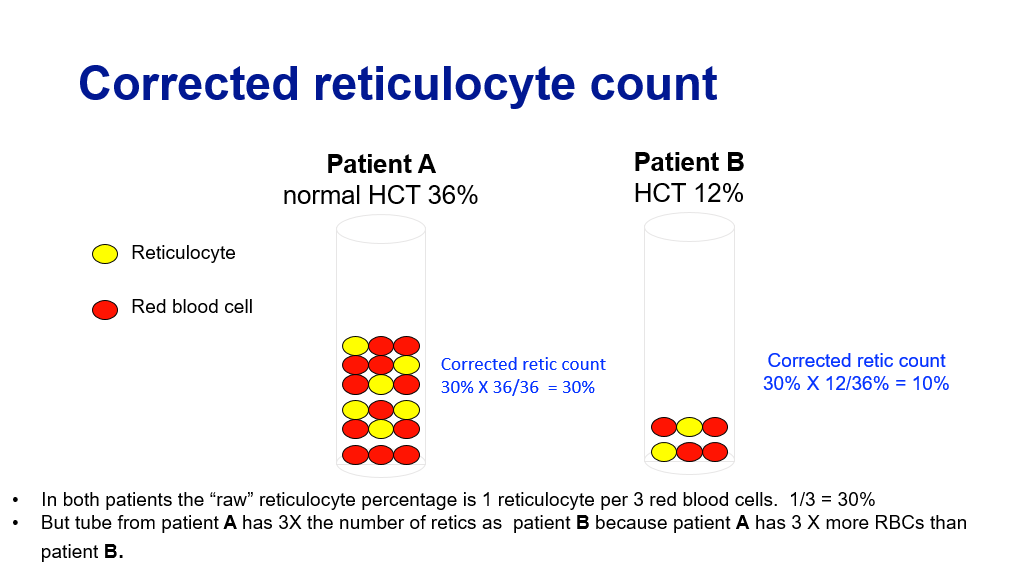
Corrected reticulocyte count = % Reticulocytes X Patient’s HCT/Normal HCT
Absolute reticulocyte count = % Reticulocytes X RBC number X 100.
—Note that a normal absolute reticulocyte count is about 20,000-60,000 retics/ul.
—A corrected reticulocyte count is considered increased if it is at or above 3%.
If a patient has anemia and has a corrected reticulocyte count less than 3%, then they aren’t producing reticulocytes appropriately for their degree of anemia, and an unproduction problem is generally the cause of anemia in that situation.
Let’s now go through the 5 lab steps for evaluating anemia again:
1. Are the RBCS produced appropriately? The first step in evaluating a patient with anemia is to determine the corrected reticulocyte count. If the corrected retic count is above 3%, then the patient is making reticulocytes appropriately for the degree of anemia.
2. If the corrected retic count is low, are the RBCs maturing appropriately? (What is the size of the RBCs?) If the reticulocyte count (corrected) is low (less than 3 %), then this is an anemia with decreased RBC production (a bone marrow “factory” production problem). In this setting, knowing the RBC size (MCV) will help guide as to the cause.
-
- Decreased production anemias with microcytic red blood cells (low MCV, <80 fL): iron deficiency, thalassemia, sideroblastic anemia, some anemias due to inflammation (generally, in setting of microcytic anemias, the problem is related to heme or globin production due to deficiencies)
- Decreased production anemias with normal sized RBCs (normal MCV, 80-100 fL): Primary bone marrow failure (such as aplastic anemia or myelophthistic process), anemia of inflammation, renal disease (lack of epo to drive RBC production), acute bleeding.
- Decreased production anemias with enlarged (macrocytic) RBCs (high MCV, >100 fL): Megaloblastic anemia (due to B12 or folate deficiency), Myelodysplastic syndrome, anemia due to liver disease, hypothyroidism, Pure Red Cell aplasia.

Small pearl: If the MCV is above 120 fL, then the anemia is almost always megaloblastic—due to B12 or folate deficiency.
3. If the corrected retic is high (3% or higher), do the labs support hemolysis? If the reticulocyte count is not low (is at or above 3%), then we are looking at an anemia associated with increased RBC production by the bone marrow. There are really 3 reasons to see this:
-
- Hemolysis
- Blood loss with sufficient folate, iron, and B12
- Marrow recovery from a recent insult
So, step 3, accounting for these issues, aims to look for hemolysis if the reticulocyte count is high. Hemolysis is destruction of red blood cells and can occur either intravascularly or extravascularly. Hemolysis can be detected by doing additional work up, which will either rule in or rule out hemolysis:
- Haptoglobin level: Haptoglobin is a protein mainly synthesized in the liver and is predominantly found in plasma. Haptoglobin(Hb) binds to free hgb (which is released from RBCs when they are being destroyed in the body in the process of hemolysis) and the Hb-hgb complex is then cleared from the circulation by macrophages. Hence, the haptoglobin will be low in the setting of hemolysis (and, yes, it can also be low in setting of significant liver disease, but we’ll worry about this in another block). A haptoglobin of </=25 has a sensitivity and specificity for hemolysis of 83% and 96%, respectively. (Marchand A. et al, JAMA. 1980:243(19).
- Lactate dehydrogenase (LDH): LDH is a cytoplasmic enzyme in many tissues. It is released from RBCs when they are destroyed, and hence LDH is elevated in the setting of hemolysis.
- Unconjugated bilirubin: You will learn more about bilirubin and its unconjugated vs conjugated form in a future block. Suffice it to say that Unconjugated bilirubin is a breakdown product of hemoglobin and will be increased in hemolysis. Unconjugated Bilirubin is also called Indirect Bilirubin by some labs. (You don’t need to now details about unconjugated vs conjugated bilirubin for this block, and you will learn more about this later.) Unconjugated bilirubin is less soluble than conjugated bilirubin, so in the setting of hemolysis, this results in an increased risk of bilirubin gallstones.
- Total bilirubin: In the real world, unconjugated bilirubin is not measured. It is calculated as the difference between total bilirubin and conjugated bilirubin measurements. So for a work-up of hemolysis the total bilirubin is measured. If it is high, then the conjugated bilirubin is measured, and the unconjugated bilirubin is calculated.
- Peripheral blood smear review: If labs support hemolysis, one generally jumps to blood smear review next (and then goes back to assess other cell lines) to see if the Peripheral blood smear shows red blood cell fragments, also known as schistocytes. Do we see spherocytes? Other red cell abnormalities?
4. Are the other cell lines involved? Here we want to know what is going on with the WBC, white cell differential, and platelet counts that are measured on the CBC. If multiple cell lines are effected, could reflect a more diffuse bone marrow process, such as infiltrative disease, leukemia, etc, or potentially a process impacting multiple cell lines, such as advanced liver disease, often associated with reduced platelets and WBCs, due to low TPO and splenic sequestration.
5. Look at the peripheral blood smear. The appearance of the RBCs will give us many clues as to the cause of the anemia. Here are some examples of what you may see on a blood smear that may help you sort out a cause for anemia.
- RBC fragments (schistocytes) or spherocytes: Hint at hemolysis
- Small RBCs with central pallor hint at iron deficiency.
- Target cells hint at thalassemia
- Dimorphic population (microcytes and macrocytes) with sideroblastic anemia
- Low/absent platelets and low/absent WBCs in setting of anemia indicate a factor (bone marrow production) problem or possibly liver disease with portal hypertension and splenic sequestration of cells (you’ll learn more about this in another block)
- Hypersegmented neutrophils are seen in B12 or folate deficiency.
We will now move on to discuss systemic iron homeostasis. It is important to understand iron homeostasis, as the leading cause of anemia, by far, is iron deficiency.
Required video on Canvas: “Primer on Iron Homeostasis”: The slides from this talk are also made available to students on canvas – See day 11, session 1, pre-class required material.
Systemic Iron Homeostasis
Systemic iron homeostasis is maintained by regulated iron uptake and storage. Note that the body has no regulated mechanism to get rid of iron, it can only regulate uptake of iron. (In other words, if for some reason the body gets too much iron, it has no way to get rid large amounts of iron naturally.) The human body has about 4 grams of iron. Most of this iron (about 2 grams) is found in the hemoglobin contained in mature and developing red blood cells. In regards to the rest of the body’s iron stores, about 600 mg is stored in macrophages, and 1.4 grams are stored in the liver as ferritin and in other tissues. The key to maintaining systemic iron supply and homeostasis lies in the regulation of adequate plasma iron pool where iron circulates bound to transferrin and is available for various needs (bone marrow to make red blood cells, liver for storage, or other tissues). Every day about 25 mg of iron is recovered by macrophages which phagocytose senescent (aging) red blood cells and returned to the plasma pool to maintain normal daily red blood cell production. Under normal circumstances the daily losses of iron from the body is small, only 1-2 mg (generally iron is lost through gut sources or menstrual loss). This normal iron loss is readily replaced by the absorption of similar amounts of iron in the diet.
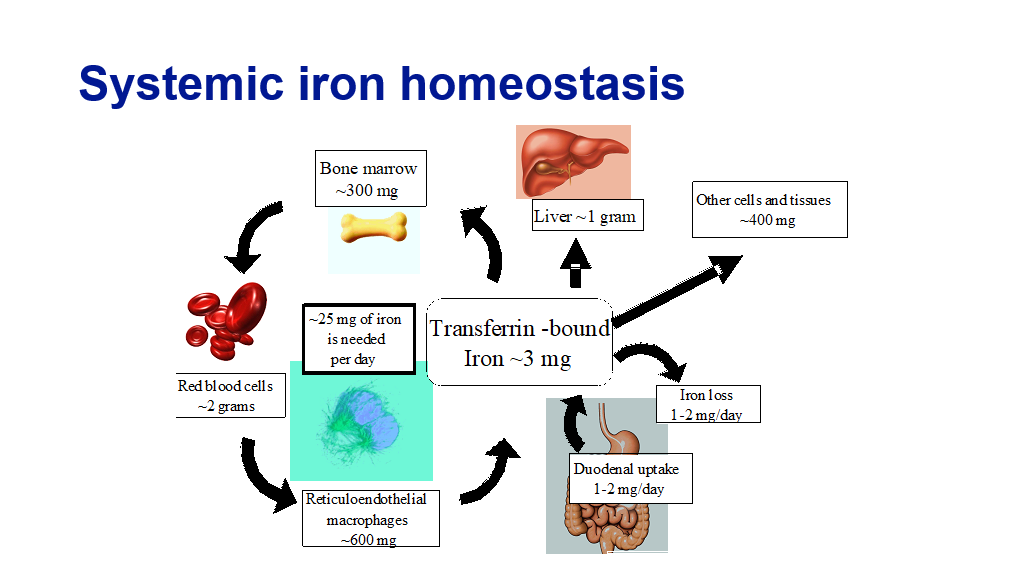
Let’s briefly review how iron gets into the body, is stored, and is utilized. Remember, Iron excretion is minimal. This slide is a good summary, and then we’ll have more details below.
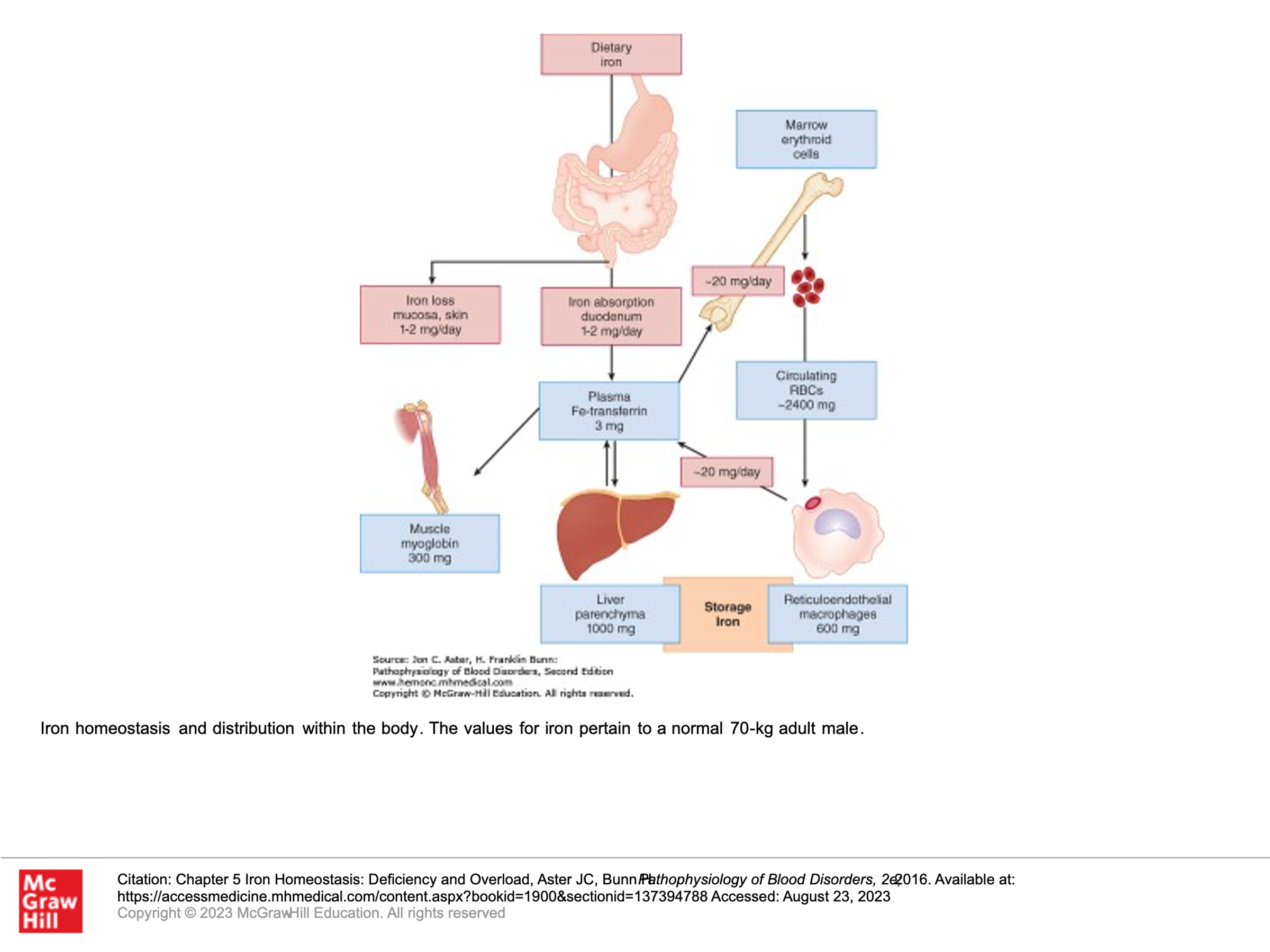
Iron absorption:
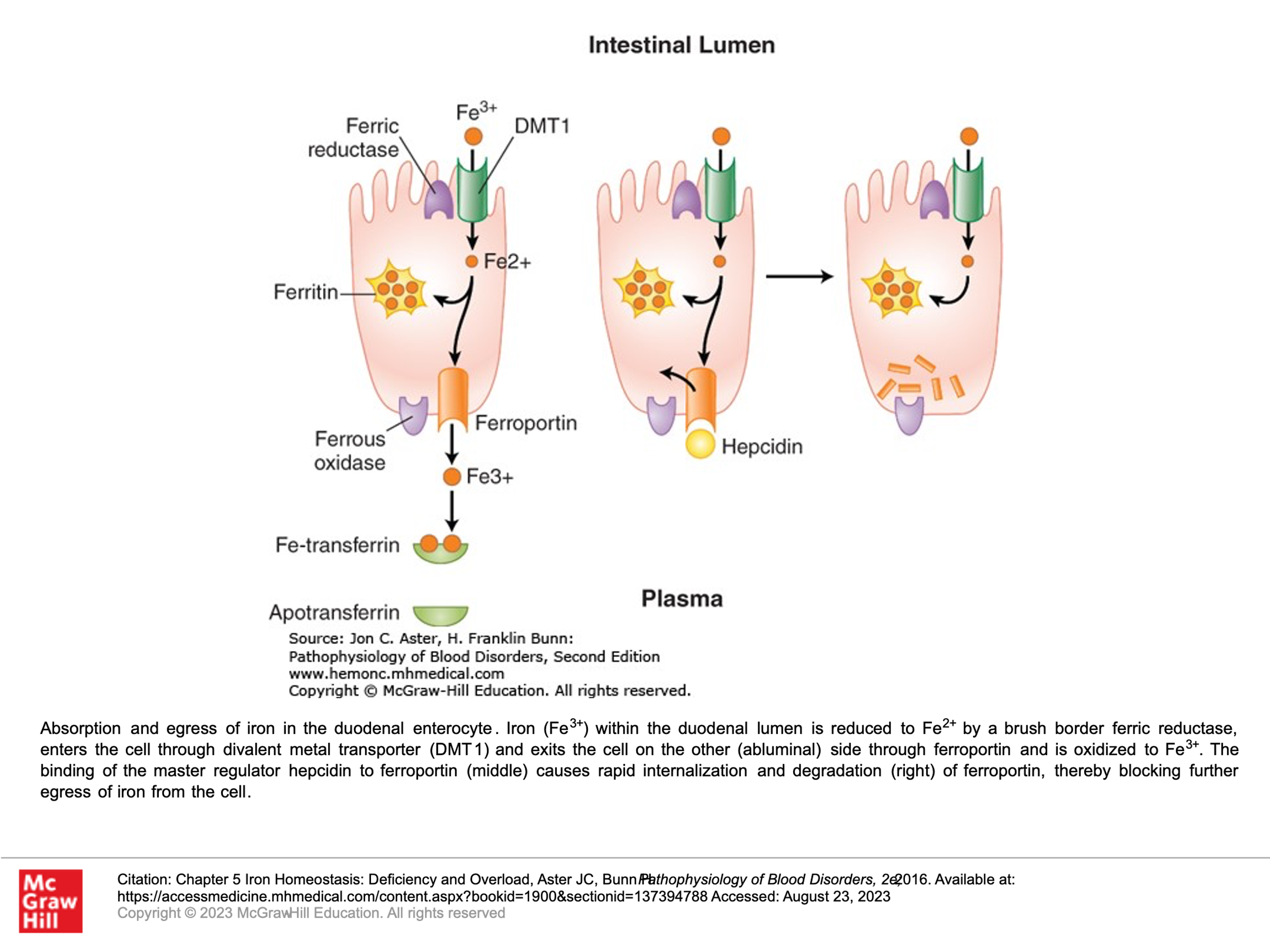
Iron from our diet is absorbed in the small bowel, primarily by the villous tips of the duodenal enterocytes. A ferrireductase at this site reduces the iron from its ferric form (Fe3+) to its Ferrous form (Fe2+), allowing the iron to then enter the cell through a luminal transmembrane channel called the divalent metal transporter (DMT1). Some of the iron that enters the small bowel enterocyte may be stored within a multimeric protein cage called ferritin. Ferrous iron that isn’t stored in the enterocyte exits the cell through the only known cellular iron exporter, a transport protein called ferroportin, which is found within the plasma membrane on the basolateral side of the enterocyte. Once the iron is exported from the enterocyte, it is quickly oxidized back to it’s ferric (Fe3+) form and then binds to transferrin, which is the plasma protein responsible for iron transport in circulation, and can which can hold two iron atoms.
Another protein that is important in this pathway is hepcidin, which blocks the export of iron from duodenal enterocytes into the circulation, impairing iron absorption. Hepcidin does this by binding to ferroportin, triggering the internalization and degradation of ferroportin. If ferroportin levels go down, then iron export from the enterocyte into circulation is markedly reduced. Hepcidin is produced by the liver, and it’s expression is tightly regulated at the transcriptional level—in part this is done by iron. Thus iron stores in the body (including in the liver) adjust iron uptake from the gut to prevent the body from absorbing too much iron. In hereditary hemochromatosis this regulation is lost due to inability to upregulate hepcidin.
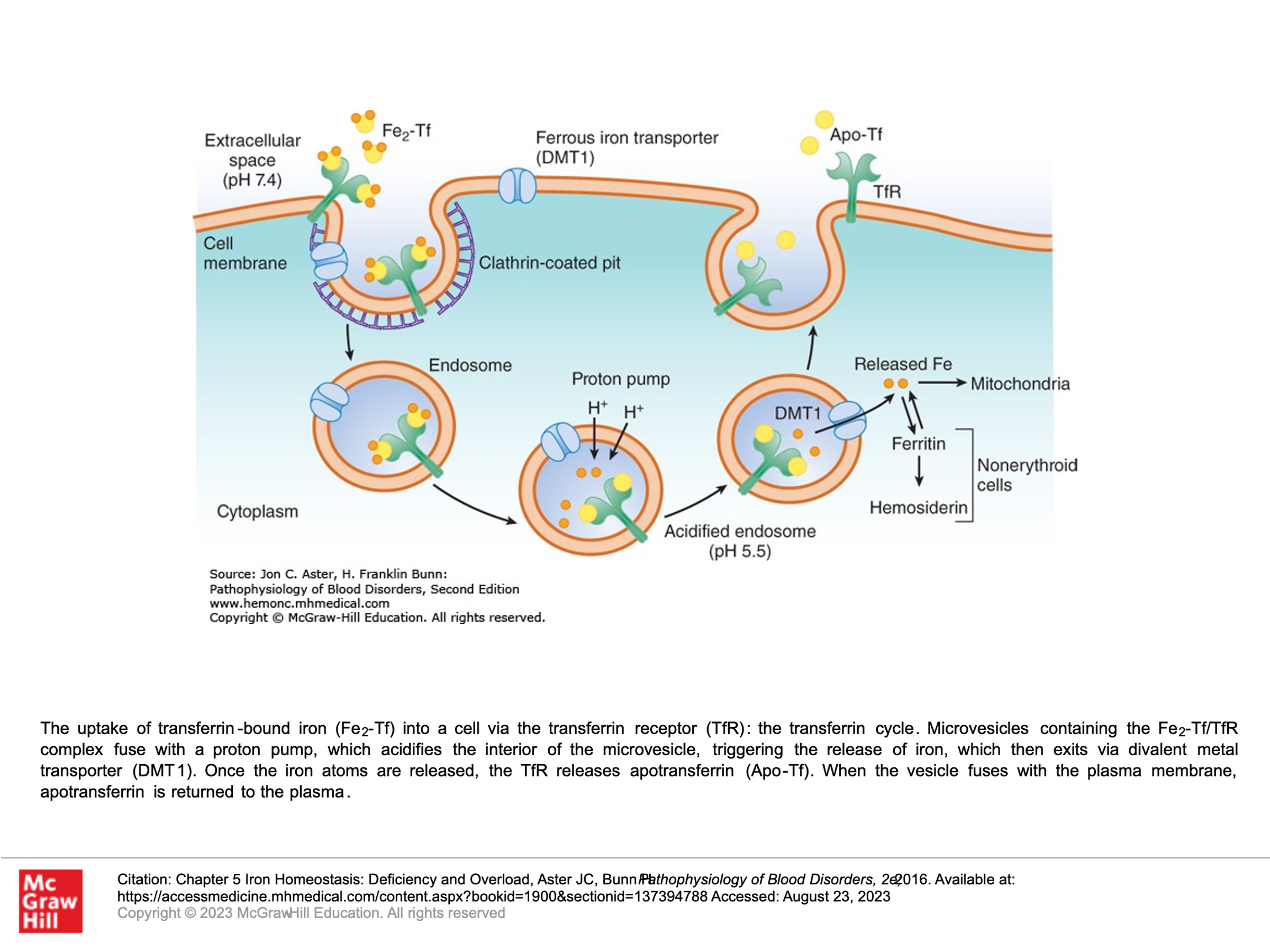
Iron transport and entry into cells:
The plasma protein transferrin binds ferric iron (Fe3+) tightly and transports iron in circulation. In doing so, it protects tissues and cells from the toxicity of free iron during its transport in plasma. Transferrin picks up iron either from duodenal enterocytes or from macrophages. Macrophages accumulate iron recycles from hemoglobin in senescent cells. All cells other than duodenal enterocytes take up iron via the binding of transferring with iron (Fe-transferrin) to transferrin receptors on the plasma membrane of those cells. As you can imagine, erythroid precursor cells that produce high levels of hemoglobin express much higher levels of transferrin receptors than do other cells. Transferrin molecules that have 2 molecules of iron have a much higher affinity for the transferrin receptor on cells than transferrin that contains only 1 molecule of iron or no iron. When Fe-transferrin binds the transferrin receptor, the complex is rapidly internalized by endocytosis into microvesicles called endosomes which also contain the iron transport protein DMT1. A proton pump docks onto the endosome and acidifies its interior, thereby releasing transferrin-bound iron. The free iron is then exported via DMT1 into the cytosol, where iron is either directed to mitochondria for heme biosynthesis or stored as ferritin. Transferrin receptors freed of their cargo may then be recycled back to the cell surface.
Iron utilization in erythropoiesis:
We have previously covered how iron is utilized to make hemoglobin. See course pack material on erythropoiesis.
Iron recycling:
As red cells age, the spleen recognizes aged RBCs and takes them out of circulation (rbc senescence)—this is mainly done by macrophages in the spleen and also in the liver. These macrophages degrade hemoglobin and incorporate the released iron into ferritin, which is stored in their cytosol. Like the duodenal enterocytes, iron leaves macrophages via ferroportin and is bound to transferrin in the plasma. Normally, the flux of iron from macrophages back to the bone marrow is about 20 mg per day, much higher than the daily ingestion of 1-2 mg of iron from gut absorption. Hence, in vivo iron homeostasis relies heavily on recycling of iron from senescent RBCs via macrophages and then delivering this to bone marrow through circulation. This system can be impacted by inflammatory cytokines. Certain inflammatory cytokines upregulate the expression and secretion of hepcidin from hepatocytes, blocking no only iron uptake by enterocytes but also the efflux of iron from macrophages. (This is the cause of anemia due to chronic inflammation.). Recall that hepcidin not only blocks release of iron from duodenal enterocytes, but also release of iron from macrophages (leading to iron sequestration). This iron is thus not available for erythropoiesis.
Iron storage:
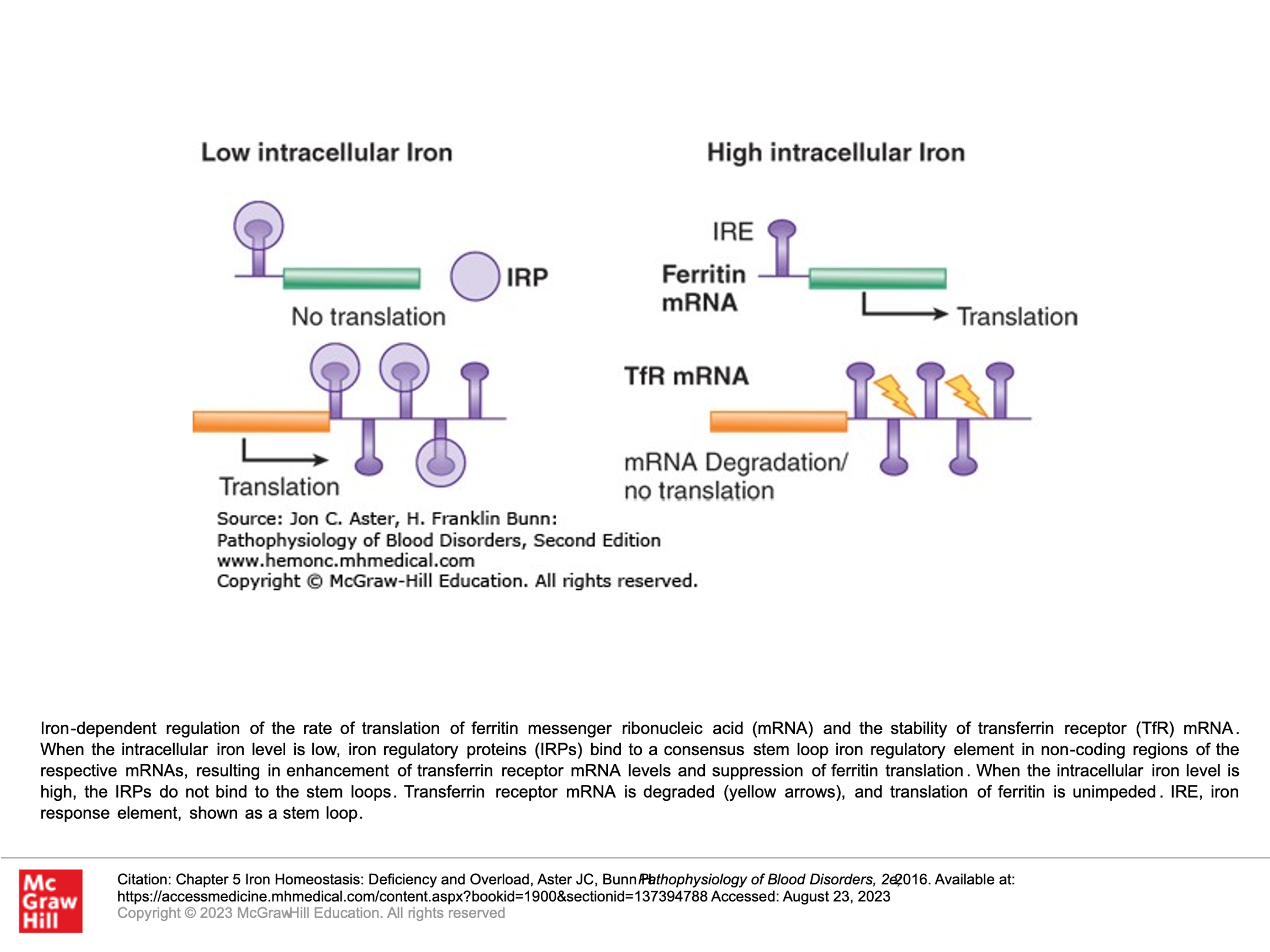
Because free Iron (Fe3+) is toxic, it must be stored in ferritin. Ferritin is a protein that assembles into a multimeric 24-subunit cage surrounding an inner core of iron hydroxide. Ferritin complex is able to store 4,500 iron atoms. The production of ferritin is tuned to the cell’s need to store iron. When iron is scarce, ferritin protein production is halted. Essentially, iron regulates the rate of translation of ferritin messenger RNA, hence the correlation of ferritin production with need for iron storage.
We measure iron by measuring the following measures (Iron studies):
- Serum iron (measures amount of iron in the serum). Normal is about 50 to 150 ug/dL. Note that this measure will vary during the day depending upon iron intake, etc. for example after dietary or supplemental iron, the serum iron would increase but this value does not reflect total iron stores in the body. The serum iron basically measures the total amount of iron in the blood, most of which will be iron bound to transferrin.
- Total iron binding capacity (TIBC): Iron is transported on proteins in plasma, the most common is Transferrin. The binding capacity is the amount of receptors on an iron binding protein not saturated with iron. A high TIBC is seen in the setting of iron deficiency. The TIBC is measured by drawing blood and measuring the maximum amount of iron it can carry, which indirectly measures transferrin since transferrin is the most dynamic iron carrier. It is apparently less expensive to measure TIBC in the blood than a direct measurement of transferrin.
- Iron Saturation (or Percent Saturation): This generally refers to Transferrin Saturation.
- Transferrin Saturation: Normally, transferrin is 1/3 saturated with iron. In setting of iron deficiency, the transferrin saturation (amount of transferrin saturated with iron) will be low eg <15%. Transferrin saturation is measured as a percentage. It is the value of the serum iron divided by the total iron binding capacity.
- Transferrin: Transferrin is the main plasma protein for transport of iron. Its concentration correlates with the total iron-binding capacity of serum. For diagnosis of iron depletion states, transferrin and total iron binding capacity can be used interchangeably. Transferrin is synthesized primarily in the liver. In otherwise healthy individuals with iron depletion states, transferrin levels in serum rise due to an increase in synthesis by the liver. High levels can be seen in pregnancy and during estrogen administration. Decreased transferrin may be seen in chronic liver disease, malnutrition, and protein loss. It is important to note that transferrin is decreased in malignancy and in both acute and chronic inflammation. Transferrin is a negative acute phase protein, meaning expression goes down in the setting of inflammation.
- Ferritin: A measure of stored iron. Normal is about 45-340 ng/dL. very low values are diagnostic of iron deficiency while elevated levels can reflect iron overload, but also liver inflammation, or other forms of inflammation/infection since ferritin is an acute phase protein.
Note: Transferrin: This is an iron transport protein. It is normally 1/3 saturated with iron.
Note: Transferrin Receptor: This is highly expressed on erythroid cells, hepatocytes, and placenta. This receptor binds transferring-iron complexes that are then endocytosed into the cell. Iron is released into the cell. Apotransferrin and transferrin receptor are then recycled to the cell surface.
Note: Ferritin: This is a large globular protein that binds iron. Ferritin reflects body iron stores, and a measure of <10 ug/L is diagnostic of iron deficiency. Ferritin is found in liver and in plasma. Of note, ferritin is also an “acute phase reactant”, meaning that levels increase with liver injury or inflammation.
Iron Deficiency Anemia
Clinically, iron deficiency anemia is by far the most common cause of anemia. The clinical history is extremely important in evaluating all cases of anemia, including anemia due to iron deficiency….
Typically, the patient history (clinical history) is unremarkable in the setting of iron deficiency anemia, unless patient recalls heavy bleeding such as GI bleeding or menses. Obviously, the first question to ask a patient is “Have you had any heavy bleeding/frequent bleeding?” Patients may not always know they have had bleeding if they are having occult bleeding. Folks who menstruate don’t typically associate this with an “are you having any bleeding” question—so it is important to ask about menstruation in addition to other episodes of bleeding. It is important to ask folks who menstruate about the frequency and severity of their menses, etc., as part of the clinical history gathering of any anemia work up. Fatigue is a common complaint with all anemias. A classic symptom with iron deficiency is the craving of crunch things such as ice, bricks (most patients won’t volunteer the information that they crave eating ice, bricks or rocks/dirt, so it is important to ask). Sometimes, one can see clinical findings of iron def. The obvious one is pallor. Less obvious findings with iron deficiency include curved nails—they look like spoons that could hold liquid, also called koilonychia. Plummer-Vinson syndrome is a classic finding in significant iron deficiency characterized by difficulty swallowing, iron def anemia, glossitis, cheilosis, and esophageal webs.
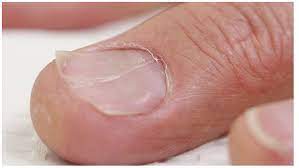
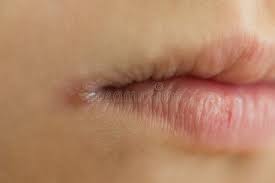
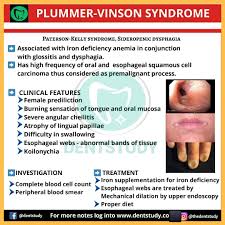
After the clinical history is taken, laboratory assessment, using the 5 steps above, will help guide you to a diagnosis of iron deficiency anemia. This will present as an underproduction anemia with low retic count, prompting you to pursue additional tests to evaluate for iron stores.
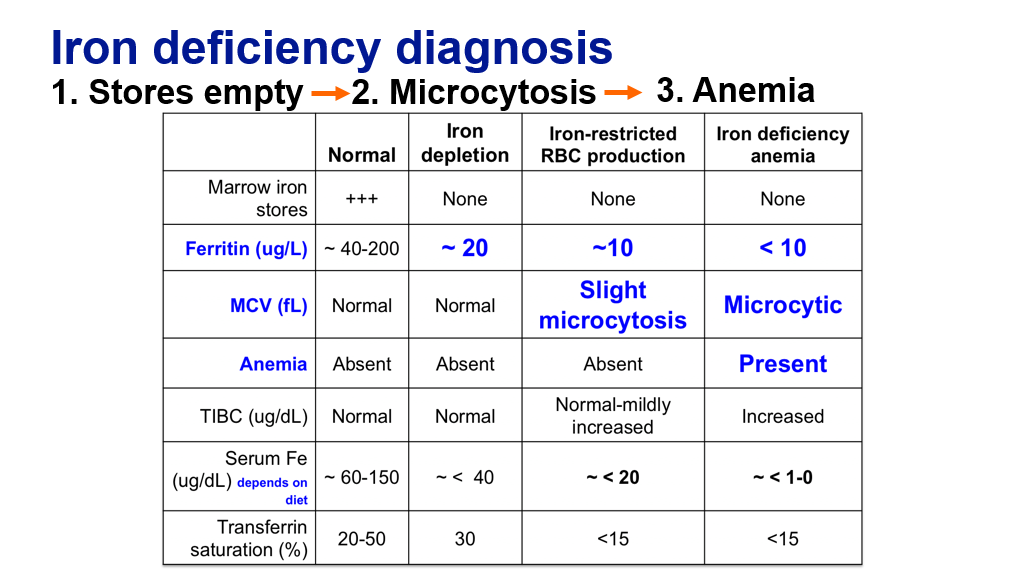
Iron studies can be helpful to define the degree of iron deficiency. There are various changes in the iron study labs as one progresses from developing iron depletion to frank iron deficiency anemia. Note the order of changes in iron deficiency leading to iron deficiency anemia.
- Iron stores are depleted prior to development of microcytosis and anemia.
- Red cells become microcytic (low MCV) generally before anemia develops in the setting of iron depletion.
- Anemia develops after iron stores are depleted and after the MCV drops.
- TIBC increases as iron binding capacity increases.
- Platelets also rise with advanced iron deficiency (not shown on table), mechanism not well defined but may reflect in part a shift to megakaryocyte production from megakaryocyte-erythroid progenitors in the marrow in the setting of iron restriction.
Treatment of Iron Deficiency
Treatment of iron deficiency: Give Iron…. Okay, it’s not quite so simple (but close):
To treat iron deficiency, it is important to understand how iron is absorbed in the gut, which we just covered in detail above. Here is a succinct summary below with some added pearls:
- Western diet contains about 12-18 mg of elemental iron.
- Ferric (3+) iron is solubilized and chelated in the acidic stomach.
- Uptake of solubilized/chelated iron occurs in the duodenum and upper jejunum. 10% of non-heme iron is absorbed. >30% of heme-iron is absorbed.
- Iron supplements should not be given with food, and particularly NOT with calcium supplements—as these interfere with iron absorption.
- Iron supplements should be given 2 hours before or after antacids (that is because iron is absorbed better in an acid environment).
- Ascorbic acid (vitamin C) increases iron absorption, so have patients take their iron with OJ.
Treat iron deficiency with Iron Replacement Therapy:
- Suggest iron-rich foods. This can be one way to treat mild iron def, particularly if patients are compliant with diet and are motivated because oral iron supplements bother their stomach.
- Oral iron supplements
- Ferrous sulfate (65 mg elemental iron/325 mg tablet or in liquid form). Other forms/doses of iron are also available if the patient doesn’t tolerate this dose.
- Absorption is improved if the iron supplement is taken every-other-day (vs daily). oral intake leads to a feedback upregulation of hepcidin for 24-48 hours.
- Side effect is constipation (related to iron content) and some people have nausea.
- IV iron preparations such as Iron sucrose or Iron dextran. (You are not responsible to know the different formulations/doses of IV iron). Main side effect is allergic reactions which is much less common with newer formulations. The risk of allergic reaction varies with the iron preparation. IV iron bypasses oral absorption so is not associated with constipation or other gastroinestinal symptoms.
- Red blood cell transfusion: If all else fails and patients can’t tolerate oral iron or have malabsorption and have allergic reactions to IV iron, then RBC transfusion to treat anemia is a last resort.
Anemia Due to Hemolysis
Anemia due to hemolysis is far less common than iron deficiency anemia. Hemolysis is the most common cause of an increased production anemia (anemia with high corrected retic count) that is not due to acute bleeding. It is important to recognize hemolysis in the clinical setting. Patients typically present with fatigue/symptoms due to anemia. Patients do not report significant bleeding. The anemia can be profound. Again, the lab steps to evaluate Anemia are utilized. Once an elevated retic count is found, then hemolysis labs are done. It is important to be familiar with Hemolysis labs: LDH, Total bilirubin (or Unconjugated bilirubin, or both), Haptoglobin.
If the LDH and total bilirubin are high and the Haptoglobin is low, hemolysis is highly likely.
Once we have determined that hemolysis is occurring, the next step we need to do is to figure out what kind of hemolysis: Fragmentation hemolysis vs Autoimmune hemolysis. The peripheral blood smear is a very useful test to tell us this.
Fragmentation hemolysis, which is typically seen in the setting of DIC, TTP, and traumatic hemolysis, will be evident on a blood smear by the presence of red cell fragments, called schistocytes.
Autoimmune hemolysis related to antibodies binding to red blood cells and causing them to be removed by the spleen generally doesn’t cause red blood cell fragmentation. In warm auto immune hemolytic anemia, due to IgG autoantibodies, hemolysis can cause red blood cell spherocytes to appear in the peripheral blood. Warm auto antibodies are IgG and can have specificity to Rh antigen on RBCs. Warm auto immune hemolytic anemia is characterized by extravascular hemolysis. Antibody coated RBCs are cleared through the spleen, and due to opsonization can form spherocytes as they passage through the spleen, leading to reduced RBC survival, and elevated reticulocyte count.
Autoimmune hemolysis due to cold auto antibodies are IgM and react against the I antigen on RBC. IgM antibodies or pentameric in structure and can lead to RBC clumping/agglutination appearance on the peripheral smear.
In hemolysis the MCV may be elevated since reticulocytes are increased in % and absolute number, and are of higher volume than mature RBCs (150 fL compared to 85 fL)
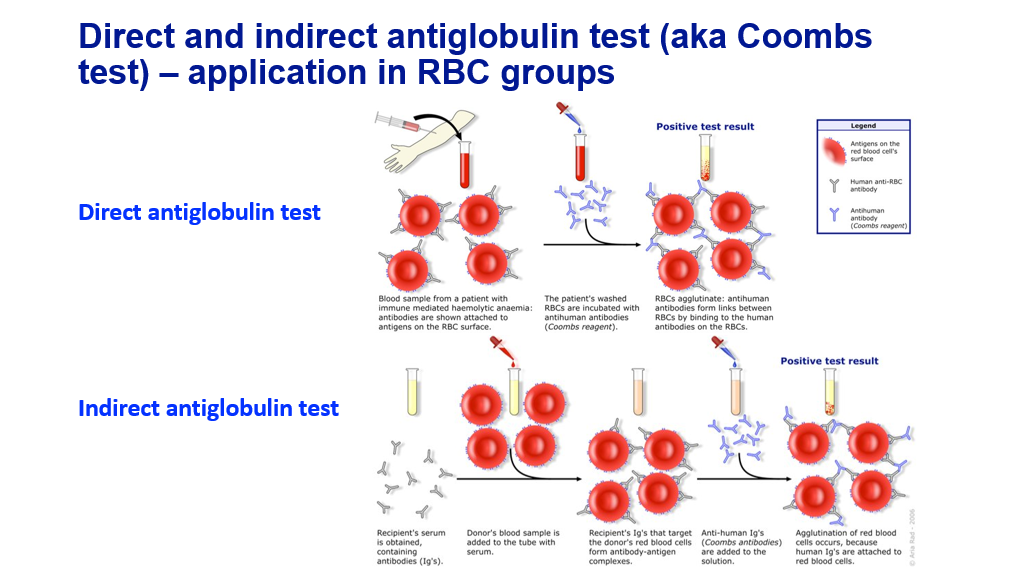
The Coombs test can be used to detect whether hemolysis is due to autoimmune processes.
So, if you find you have an increased production anemia (anemia with elevated corrected retic count of 3% or more), then you should check labs to evaluate for hemolysis (LDH, bilirubin, haptoglobin). If those labs support hemolysis, you next want to look at the blood smear. You also may want to do Coombs testing (Direct Coombs testing is also known as a Direct Antiglobulin test), particularly if the blood smear doesn’t show significant fragmented RBCs (fragmented RBCs are generally not seen in autoimmune hemolysis and are pathognomonic for fragmentation hemolysis).

Coombs testing: Direct Coombs (direct antiglobulin) test and Indirect Coombs (Indirect antiglobulin) test.
A direct antiglobulin test tests the patient’s red blood cells to see if there are antibodies bound (auto-antibodies to the patients own red blood cells). This is the test that is typically done when one suspects a patient has an autoimmune hemolytic anemia. Blood is drawn from the patient. The red blood cells are washed so that any non-specific antibodies not bound to the cell fall away from the cell. Autoantibodies that bind to the cell will stay on with washing. The patient’s washed RBCs are then incubated with anti-human antibodies that will bind to human antibodies on the RBCs (this is the Coombs reagent). These anti-human antibodies will then cause red blood cells that have autoantibodies on them to agglutinate. This test would be abnormal in both warm and cold agglutinin hemolytic anemia, leading to a positive DAT result.
An indirect antiglobulin test is generally done when one is crossmatching a patient to look for compatible red blood cells—test if donor red blood cells that will be transfused are compatible for the patient. The patient’s serum is obtained, which contains antibodies. The donors red blood cells are then incubated with the patient’s serum. If the patient has antibodies that will react to the red blood cells, then these antibodies will bind to the red blood cells. Antihuman antibody (Coombs reagent) is then added to the solution. If the patient’s antibodies have bound to the red blood cells/reacted to them, then the donor cells will agglutinate after they have been exposed to the patient’s serum and then the anti-human antibodies. If this occurs, then the donor’s red blood cells are not compatible to be given to the patient. These are pre-formed allo-antibodies in the patients serum that react against donor RBCs. Students will learn more about transfusion medicine later in the block. Note that patients with sickle cell anemia have increased risk of developing allo-antibodies in part due to the need for chronic transfusions, though other factors may also play a role, which can make finding compatible red blood cells difficult for these patients.
Summary
- We have reviewed a stepwise approach to the evaluation of anemia.
- We have addressed how to calculate the reticulocyte count and the concept of underproduction vs increased-production anemias.
- In the setting of increased-production anemias, we have summarized testing for hemolysis.
- We have reviewed iron homoeostasis, as a basis to understanding iron deficiency and the labs that we utilize to assess for iron deficiency.
- We have reviewed lab assessment for iron deficiency.
- We have reviewed Iron deficiency anemia and its treatment.
- We have discussed hemolytic anemias and the broad characterization of these as due to fragmentation hemolysis vs autoimmune hemolysis.
- We have reviewed Coombs testing, which is utilized testing for autoimmune hemolysis.
Optional textbook reading: Part 1 Anemia and Red Cell Disorders
Chapter 11: Acquired hemolytic anemias (section on immune hemolytic anemias- can skip treatment section)
Authors and Contributors:
Corliss Newman, MD authored this course pack material based upon slide lecture material prepared by Sioban Keel, MD. Section on iron homeostasis written with assistance from our course textbook by Aster and Bunn, Pathophysiology of Blood Disorders, Chapter 5. Kristi Rice, MD and Nicholas Burwick, MD provided editing assistance.