
7 Blood Components, Pre-transfusion testing, and Adverse events
Student Learning Objectives:
- Describe the donor process including donor screening (self-deferral, questionnaire, and donor testing) and the different types of blood collection (whole blood and apheresis), and the risks of transfusion transmitted diseases.
- Describe the different types of blood components available for transfusion (red blood cells, plasma, platelets, granulocytes), and recognize the evidence-based indications for their use.
- Explain the reasons for requiring ABO and Rh matching prior to transfusion and analyze the basis of pretransfusion testing.
- Analyze the adverse events associated with transfusions including risk of transfusion transmitted diseases and non-infectious complications.
- Apply transfusion medicine knowledge to the process of obtaining consent for transfusion (including discussion of benefits, alternative treatment, and potential adverse events).
USMLE Content descriptors: Reactions to blood components.
Optional Textbook Reading: Part IV transfusion medicine, Chapter 25: Blood Transfusion. This 10 page chapter is a quick read and quite helpful.
Introduction:
As a physician, you will have times in your career when you need to transfuse patients with blood products. We’ll start off this day with a discussion of the different blood products available to give patients and how they are derived.
Whole blood collection and storage into components for transfusion:
As a blood donor, when you go in to donate, you give a unit of whole blood, which is about 500 ml of blood. The blood bank collects the unit from you and then promptly processes it into components for use. First the unit of blood undergoes spinning in a centrifuge at a low spin rate (a soft spin), and packed red blood cells (PRBCs) are separated from the rest of the product, which contains plasma and platelets (this product is called “platelet-rich plasma”). The platelet-rich plasma is then further spun down in a centrifuge at a higher rate of spin (a hard spin) which allows this platelet-rich plasma to be further separated into a platelet concentrate and Fresh plasma. Platelets are stored at Room Temperature. Fresh Plasma is frozen (Fresh Frozen Plasma or FFP). It can be thawed later and administered to patients who need all the derivatives in plasma (think of cases of massive transfusion, where patients need to be administered both PRBCs and FFP). Alternatively, the FFP can be further processed/fractionated into plasma derivatives such as IVIg and albumin. If FFP is thawed at 4 degrees Celsius, cryoprecipitate will precipitate out and can be centrifuged off. This cyroppt is then resuspended in a small amount of residual plasma (generally 10 to 15 ml) and is re-frozen for storage. Cryoprecipitate (aka cryo, aka cryoppt) contains fibrinogen, factor VIII, Von Willebrand factor, and Factor XIII. It is generally used as a source for fibrinogen. (There are better factor replacement products for factor VIII and VWF, so generally cyroppt is not used for this.)
Hence, from a unit of whole blood, we can obtain the following products:
- Pure Red Blood Cells (PRBCs). (Presumably we call this pure because the product is only red blood cells and a little plasma.)
- Platelets.
- FFP
- (and 5). FFP can be used as is, or it can be further processed to get cryoppt. Alternatively, FFP can be processed to get plasma derivatives such as IVIg and albumin.
“But wait,” you ask (because you are so darn smart), “what about the White Blood cells?” As you can recall, WBCs do centrifuge out in a “buffy coat” when a lab tube for a CBC is spun down. The buffy coat is composed of platelets and WBCs. In most blood collection centers, the WBCs are removed at the time of collection or at the time of blood product transfusion by a filtering mechanism, as WBCs are a cause of transfusion reactions and a mechanism for transmission of CMV (cytomegalovirus infection). In the extremely rare case where WBCs are needed for infusion to treat patients, the blood bank has a separate way of getting this type of product, and one would generally work with a hematologist and a blood bank directly in this type of exceedingly rare setting. WBC transfusion (AKA Granulocyte transfusions) are only indicated for severe and refractory fungal infection for neutropenic patients, and have risks with administration.
Thus, in total, there are 5 different products (not including WBCs) that are commonly used in medicine that are obtained from whole blood: PRBCs, platelets, FFP, Cryoppt, and plasma derivatives (IVIg, albumin, etc.).
We’ll now go on to talk about these blood component products:
Red Blood Cells (RBCs, pure red blood cells, PRBCs—we use these terms interchangeably)
Red blood cells are given to patients for certain indications only. These indications are as follows:
- To increase O2 carrying capacity to tissues
- To replace red blood cells following significant hemorrhage.
- When other treatments for chronic anemia have failed.
It is very important to only give RBCs for approved indications, and RBCs are a donated resource and can become scarce/is limited at times.
Bag of Red blood cells for transfusion
One unit of PRBCs is about 350 ml and is comprised of 200 ml of red blood cells, 100 ml of a storage solution (RBCs can be stored for up to 42 days at 4 degrees Celsius), and 50 ml of plasma.
When we administer PRBCs, it is important to match a donor with a recipient such that the donor and recipient are ABO and RhD compatible (more on this later).
In a patient with anemia, giving 1 unit of PRBCs will roughly raise the hemoglobin by about 1 g/dL. In other words, it will raise the hematocrit by about 3%.
Recommended Transfusion thresholds (at what hemoglobin measure we typically use to decide whether to administer blood) have varied over the years. However, several studies have shown that transfusions for higher hemoglobin thresholds of 9 to 11 result in adverse outcomes compared to lower hemoglobin transfusion thresholds of 7-9. Hence, we tend to favor more restrictive transfusion thresholds, and we generally use a hemoglobin cut off of 7 for transfusion in an otherwise stable patient. In plain speaking, we generally transfuse if the hemoglobin is less than 7.
Platelets:
As you recall from a prior session, platelets are involved in primary hemostasis. Hence, they are important to control bleeding. Indications for platelet transfusions include thrombocytopenia –there are different targets for transfusion depending upon the clinical situation. Platelets also can be given for platelet function disorders to replace platelets that aren’t functional—in that setting platelets are given regardless of the patient’s platelet count (as presumably the patient’s own platelets aren’t working very well and the patient is bleeding). For patients how don’t have a known disorder of platelet function, the following slide is a general guideline for when we transfuse platelets:
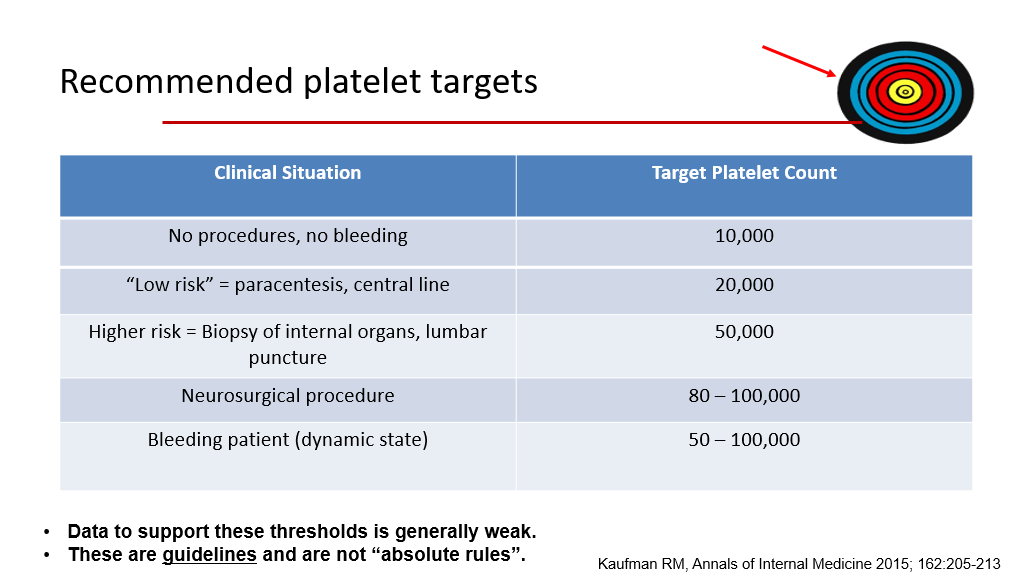
There are 2 different types of platelet products.
- Whole blood platelets: In the old days (before apheresis), we would generally give an adult about 4 to 6 units of platelets. In other words, platelets from 4 to 6 different donors would be combined together to make one adult dose of platelets. (Back in the old days, it was common to order “a 6 pack of platelets” when a patient needed a platelet transfusion.) In current times, whole blood platelets are rarely used for transfusion.
- Single Donor platelets, also known as apheresis platelets: Currently, most of the time platelets are collected from a single donor in a process called apheresis. This is different than whole blood donation. In this setting, whole blood is removed from the patient and is processed in an apheresis machine (kind of like a dialysis machine). The platelets are removed, and the rest of the blood is given back to the patient. While a donor can’t tolerate removal of more than 1 to 2 units of whole blood at a time, donors have no problem donating lots of platelets as long as their blood is returned back to them! A single donor/apheresis platelet collection collects the equivalent of 4 to 6 pooled platelet units. Hence, when transfusing a patient with apheresis platelets, we only need to order 1 unit of single donor platelets. Typically, the order we give is to transfuse 1 unit of single donor platelets or 1 unit of apheresis platelets.
Currently, about 95% of platelets that are administered to patients are apheresis (single donor) platelets.
Platelets are generally stored at room temperature, about 20-24 degrees Celsius, because cold induces clustering of von Willebrand factor receptors on the platelet surface and morphological changes of platelets, leading to enhanced clearance of platelets/reduced platelet survival. Platelets need to be gently agitated as they are stored. The shelf life is about 5 to 7 days, depending upon the storage container used. Because they must be stored at room temperature, there is a higher risk for bacterial contamination for platelets, which is part of the reason they have a shorter shelf life.
Single Donor platelets hanging for infusion into a patient.
Granulocytes
As noted above, WBCs are not generally stored from whole blood. However, in rare instances, granulocytes (neutrophils primarily, but granulocyte preparations also contain basophils and eosinophils) are needed to treat severe and refractory fungal infections in neutropenic patients. This is rarely ever utilized in routine practice. However, if needed, they are collected from a donor who is given G-CSF (granulocyte simulating factor) to first raise their WBC. The granulocytes are then collected via an apheresis procedure (similar to what we discussed for platelets above). 1 unit of granulocytes contains about 1 x 1010 (10 billion) granulocytes. The shelf life of granulocytes is only 24 hours, so they are generally collected and then immediately administered to the patient. You can expect significant transfusion reactions with administration of granulocytes, so this type of product is usually only administered as a last-ditch effort in the setting of a severe fungal infection in neutropenic patients. This is NOT used to prevent infections, as there are many risks to giving granulocytes.
Bag of granulocytes for patient infusion:
Plasma (also called Fresh Frozen Plasma or FFP)
FFP is indicated when patients have documented multiple factor deficiencies AND have active bleeding OR have a need of an urgent procedure and have an INR of 2 or higher. (This type of scenario is not uncommon in patients with severe liver disease.)
FFP is also indicated to quickly reverse the effects of warfarin or to replete vitamin K dependent clotting factors in the setting of vitamin K deficiency when such patients have significant bleeding OR have need of an imminent medical procedure.
(Note, in a patient with severe bleeding who is on warfarin, instead consider reversing the effects of warfarin by giving Prothrombin complex concentrates instead of plasma—more on this later.)
FFP is not the preferred treatment for isolated factor deficiencies. It is not used for volume expansion (albumin is the treatment of choice for this). It is not used for low antibodies (IVIg is the preferred product to treat hypogammaglobulinemia in patients who have recurrent infections and low antibody levels).
Image of a bag of fresh frozen plasma for infusion in a patient.
We have covered the different blood product components and discussed how they are utilized. We now move on to discuss which blood products require compatibility testing for patients/potential recipients and how we go about doing such testing. We will first discuss blood groups.
Blood groups:
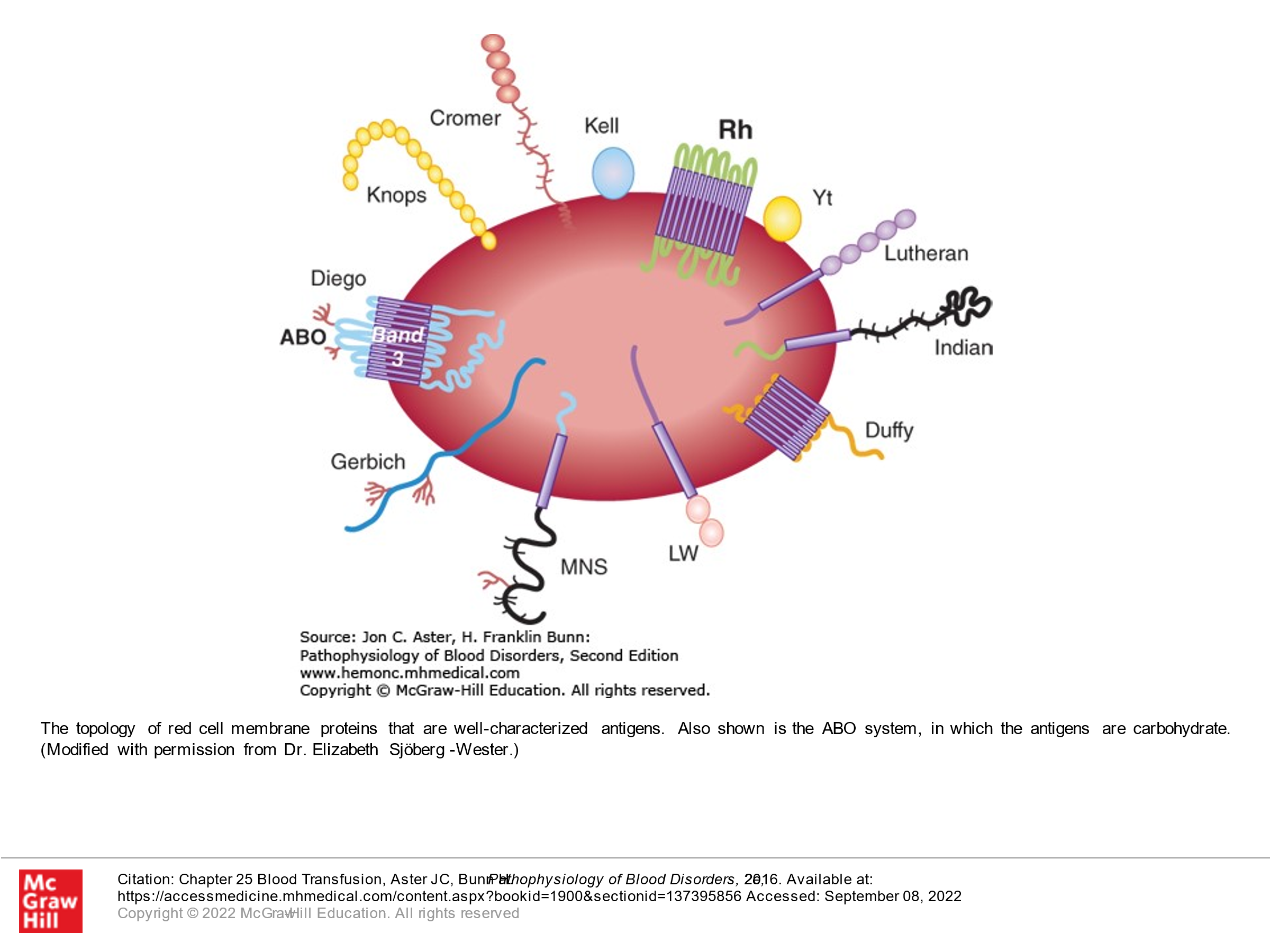
All red blood cells have antigens on the red cell surface that are genetically determined. There are 30 genetically determined blood group systems (a group is a specific antigen and its variants) resulting in over 300 different antigens. These are important, as many of these are immunogenic such that if RBCs with certain antigens are transfused to a recipient whose immune system sees these antigens as foreign, the recipient can generate antibodies to these antigens.
The best known blood group is the ABO group, which determines our blood type of A, B, O, or AB.
The second best known blood group is the Rh system (Rhesus blood group), which is composed of 2 different proteins. One of the two proteins is D. The other of the 2 proteins is either E, e, C, c, or others. The D protein is particularly immunogenic. Rh positive refers to the presence of D. Rh negative refers to the absence of D antigen on the surface of red blood cells.
Other clinically important blood groups include Kidd (Jka, Jkb), Kell (K/k), Duffy (Fya/Fyb).
ABO Blood Group
It is important to know that an antigen called the H antigen is found in all people and is present on RBCs. However, this antigen/protein in the RBC membrane is altered in many people by enzymes that bind sugar moieties to the H antigen. The ABO locus is located on chromosome 9. There are 3 allelic forms. The A allele encodes a glycosyltransferase that bonds alpha-N-acetylgalactosamine to the D-galactose end of the H-antigen, producing the A antigen. The B allele encodes a glycosyltransferase that bonds alpha-D-galactose to the D-galactose end of the H antigen, thus creating the B antigen. The O allele has a locus in chromosome 9 with loss of activity. In people with the O allele, the H-antigen is unchanged/is not bonded to a sugar moiety. Because we inherit 2 genes (one from each of our patients), our gene types can be as follows: AA, BB, AO, BO, OO, or AB. These gene types then determine our blood type: AO or AA is type A blood. BO or BB is type B blood. OO is type O blood. AB is type AB blood.

From fetal development on, individuals are exposed to A-like and B-like carbohydrate antigens from a variety of sources (not outlined in this course), and as a result we develop antibodies to A or B or both or neither depending upon what our immune system sees as foreign. These antibodies are made in the first 6 months of our life after birth. ABO is the only blood group system where antithetical antibodies are predictably present. This is summarized as follows:
-Folks with type A blood will see B-antigens as foreign and will develop anti-B antibodies.
-Folks with type B blood will see A-antigens as foreign and will develop anti-A antibodies.
-Folks with type AB blood don’t see either A or B as foreign and hence don’t’ have anti A or anti B antibodies.
-Folks with type O blood (they have only unaltered H-antigen on their RBCs) will see both A and B antigens as foreign and will have antibodies to both A and B.
These antibodies, as noted above, are derived early, and are derived even in the absence of blood transfusion. Hence, it is critical to crossmatch RBCs for ABO compatibility to avoid a hemolytic transfusion reaction.
TABLE 25-1ABO System
|
Genotype |
RBC Antigens |
Serum Antibody |
|
AA or AO |
A |
Anti-B |
|
BB or BO |
B |
Anti-A |
|
AB |
AB |
None |
|
OO |
O |
Anti-A, anti-B |
Based on the above, a person of which blood type is the universal donor (meaning their blood can be donated to anyone)? Answer: a person with type O blood (we’ll talk about Rh in a minute) is a universal donor.
Based on the above, a person of which blood type can be considered a universal recipient (they can tolerate blood from any donor regardless of whether it is type A, type B, type AB, or O)? Answer: A person with type AB blood is a universal recipient.
The Rh Blood Group:
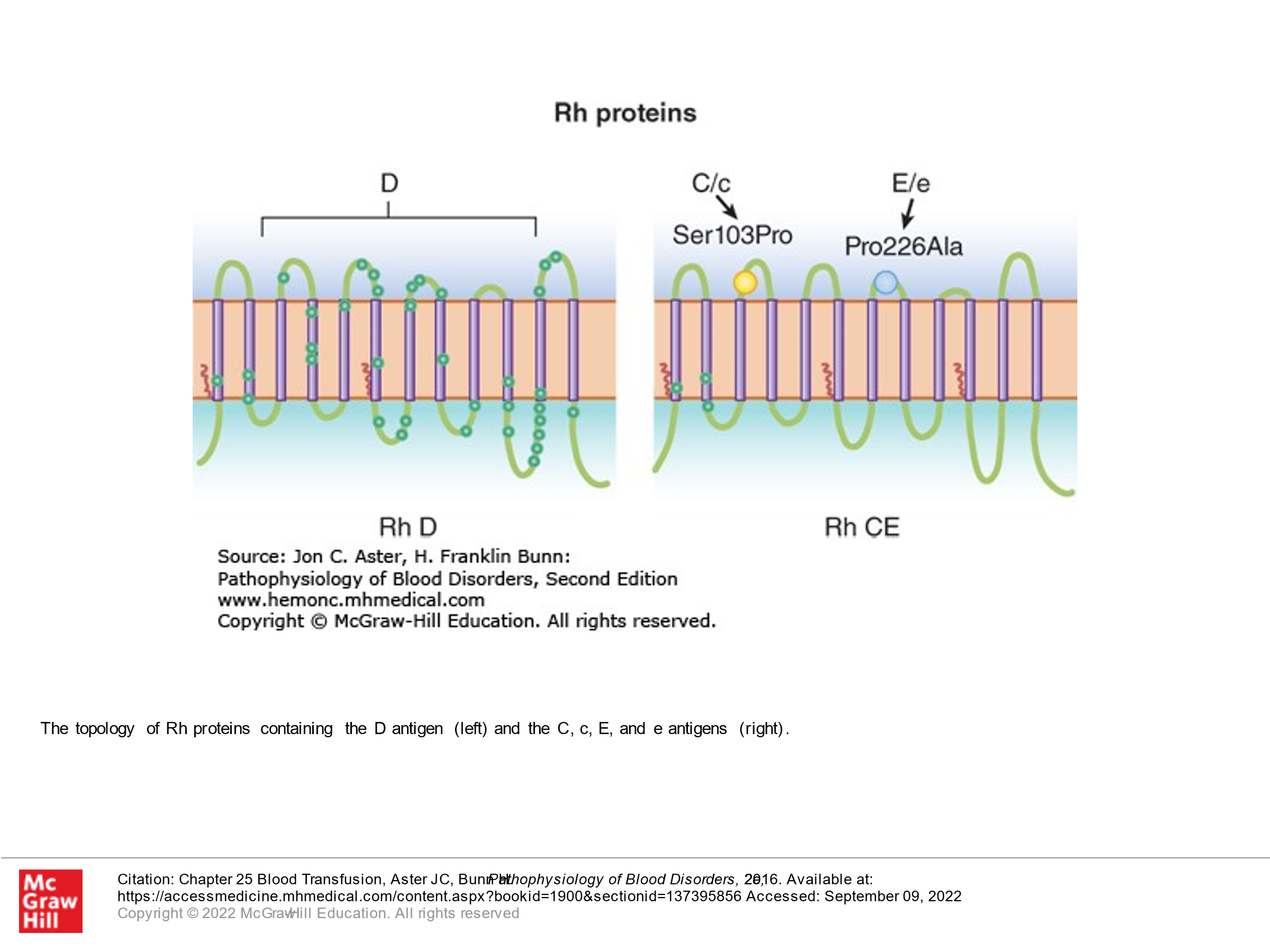
The Rh system consists of two homologous transmembrane proteins, both of which are encoded by genes on chromosome 1. These transmembrane proteins span the RBC membrane 12 times. One protein contains the D antigen. The second protein contains C/c and E/e antigens. Although we don’t naturally have antibodies to Rh proteins (unlike ABO blood group), the D antigen is extraordinarily immunogenic. About 15% of Caucasians in North America are Rh D negative. They don’t develop alloantibodies to D unless they are exposed to D-positive blood. When we talk about Rh positive and Rh negative blood, we are talking about the D antigen. Although alloantibodies can also develop against C, c, E, or e, they are much less common. Alloantibodies to the Rh system bind to RBCs, and these antibody-bound RBCs are then phagocytosed by macrophages in the reticuloendothelial system (mainly spleen). It is critical to know the Rh status of patients who are being transfused (again, this refers to D). Patients that are Rh (D) negative ideally should not receive Rh (D) positive blood, as they will make antibodies to Rh that can lead to future hemolytic reactions should they receive Rh positive blood a 2nd time in the future. Particularly important, Rh (D) negative females who develop antibodies to Rh (D) can have problems with pregnancy/resultant hemolytic disease of the fetus and newborn if they become pregnant by an Rh positive partner and their fetus/newborn is Rh positive. More on this later. However, to prevent women who are Rh negative from developing Rh antibodies (anti D antibodies) to their fetus (there is some blood exposure to the fetal blood antigens in the course of pregnancy, so this can happen in any female who is Rh negative who is pregnant with an Rh positive fetus), she is given anti-D antibody (Rhogam), which will prevent her immune system from developing anti-D anti-D antibodies. Presumably the Anti D antibodies that are given exogenously coat the D antigens on the small amounts of fetal blood that come into contact with the female’s immune system such that the person who is pregnant doesn’t sense the cells that express D and hence doesn’t develop their own antibodies. If a person develops Anti-D antibodies on their own, the Rhogam is no longer helpful and is not given.
Generally, when we transfuse blood, ABO and Rh D antigen compatible units should be given. Other antigens can be mismatched, particularly in patients who are likely only getting a one-time blood transfusion. Of note, outside of ABO/D, the next most immunogenic antigens are the remaining Rh antigens and Kell.
Blood Type and what is meant by ordering a “type and screen” vs a “type and cross”:
Blood Typing: When blood is collected, it is typed for the presence of ABO as well as presence or absence of the D antigen, and this is noted on the bag. This is called “typing the blood”. This is not a type and screen or a type and cross. This is the broad typing of the donor blood.
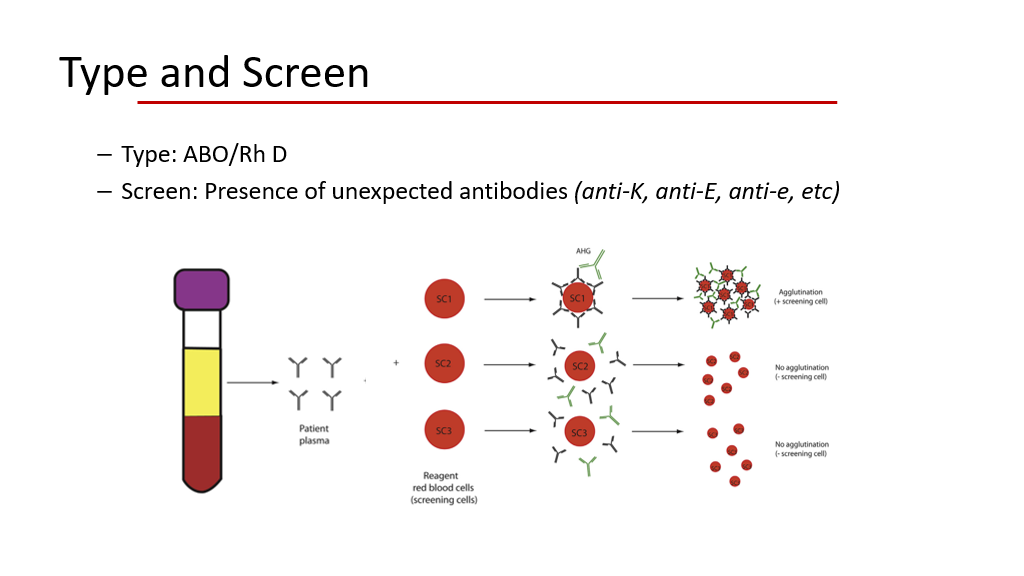
Type and Screen testing: When a patient needs blood, their blood is drawn for a “type and screen”.
- During this process, their blood is typed for the ABO and Rh D type. (That is the “type” part of a type and screen.)
- Then, their plasma is tested (aka screened) for the presence of red cell alloantibodies to a variety of blood antigens to look to see if the patient has alloantibodies to D, E, e, C, c, K, Jka antigens, etc. Generally, this is done by adding the plasma to a screening RBC sample with several known antigens. If the screen is positive, then the type of antigen the plasma is reacting to can be further narrowed down. Thus, if an antibody screen is “positive”, then even O negative blood may be a problem, because the O/D negative blood may still have other antigens such as E, e, C, c, K, Jka, etc that are causing the reaction…. In this case the blood bank does further testing to isolate the antigen of concern and then looks for specific blood donors that don’t have this antigen present who are already characterized in more detail.
The following figures depict this nicely:
- In the first figure, a reaction to screening RBC sample 1 is shown as would be seen in test tubes. There is no reaction to samples 2 and 3.
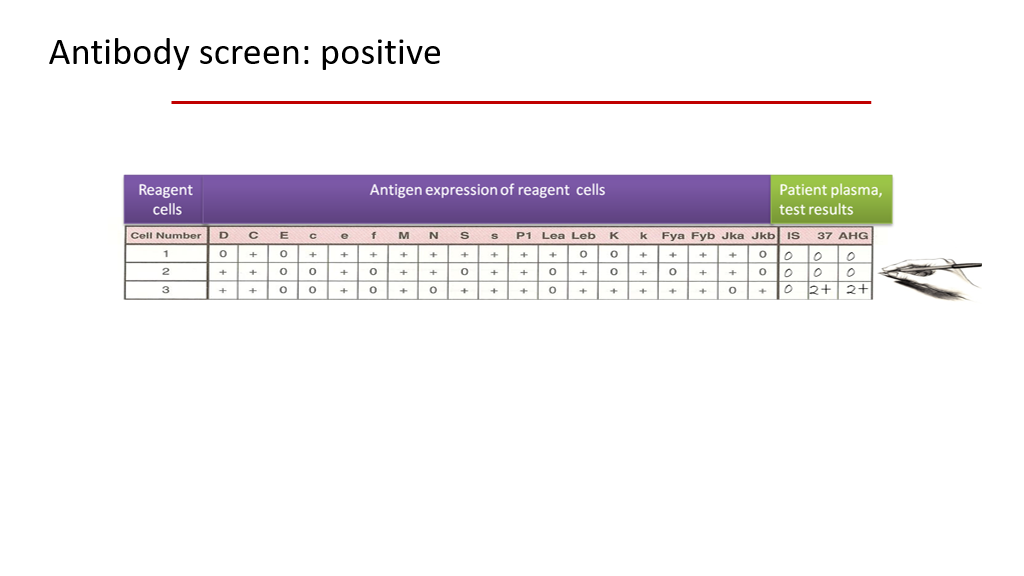
- In the 2nd figure, a table outlining the antigens found on 3 different screening RBCs is shown, along with the resulting patient plasma test results. In this slide, the patient’s plasma reacts to reagent cells in the # 3 sample.
If you look at the 3 sample reagent blood cells in the second figure below, you can see what antigens are present or not present on each of those reagent cells. Using this, you should be able to figure out that the patient’s plasma must be reacting to K or to Jkb, or perhaps they have antibodies (2 different ones) to both K and Jkb. The other antigens on Reagent cell sample 3 are ruled out because the other antigens on sample C that are present (a + sign in the table indicates that the antigen is present on the cells) also present on samples 1 and 2, and the patient’s plasma didn’t react to samples 1 and 2.
In the example in the second figure below, after the screen was done, the blood bank would then do further testing on sample RBC cells with and without K and Jkb antigens to determine what antigen the patient’s allo-antibody is reacting to. (In actuality, if the antibody screen is positive, the blood bank does further testing with a 10 cell antibody identification panel to identify the specificity of the antibody or antibodies.) They would then find a blood donor whose RBCs don’t have that antigen for the actual blood donation!
What is a Type and Cross, aka Type and Crossmatch?
This is a bit different than a “type and screen”. We often order a type and screen when we are anticipating a patient needing a blood transfusion in the future. We order a type and cross when we are planning on actually giving blood right then and there. When a type and cross is ordered, we are going to definitely transfuse blood. In this setting there are 2 pathways:
- Electronic crossmatch: The patient’s blood is typed as above for ABO and Rh and undergoes a standard plasma antibody screen. If the Antibody screen is negative, then donor blood is matched to the patient electronically based upon the donor’s RBC type/Rh. This type of a crossmatch takes about 5 minutes for the blood bank to be done.
- Serologic crossmatch: The patient’s blood is typed as above for ABO and Rh and undergoes a standard plasma antibody screen as above. If the Antibody screen is positive, then a serologic crossmatch is done. In this instance minor antigen compatibility is assessed with the 10 cell antibody identification panel against the patient’s plasma. Once a blood type/donor is identified that is compatible the plasma from the patient is also tested with a sample of the specific donor red blood cells to ensure that the donor blood cells are indeed compatible. This generally takes about 45 minutes.
Note, serologic testing where a patient’s plasma is obtained to look for antibodies to donor red blood cells is called an Indirect antiglobulin test or Indirect Coombs test. This is different than a Direct Coombs test, aka Direct antiglobulin test (abbreviated DAT). In a DAT, we are looking for RBCs that already have antibodies bound to them. Hence, a DAT is the test we do if we are suspecting hemolysis and trying to discover if RBCs are being destroyed because they have antibodies on them, and isn’t typically done as part of a blood bank procedure for crossmating. An indirect antiglobulin test/indirect Coombs test is done for serologic crossmatching, however, when selecting donor red cells to give to a patient, as we want to ensure that antibodies in the patient’s plasma don’t react to the donor red cells.
Clarification of Coombs testing (we have talked about this in both looking for autoimmune hemolysis and in the setting of blood transfusion):
Indirect antiglobulin testing/Indirect Coombs test: An indirect Antiglobulin test is done as part of an antibody screen: Plasma (containing antibodies) is incubated with Reagent red cells or donor red cells. If the antibodies target the red cells, they will bind. Anti-human Ig antibody is then added and will bind to the antibodies present on red cells and cause then to agglutinate.
Direct antiglobulin testing/DAT/Direct Coombs test: A direct antiglobulin test (DAT) is done to look to see if RBCs have antibodies on them. The patient’s RBCs are mixed with antihuman Ig antibody. If there are antibodies on the RBCs, then they will agglutinate when the anti-human Ig antibody is added. A DAT is the test that is done if a patient is transfused and appears to be having a hemolytic transfusion reaction. A DAT is also the test that is done if a patient is thought to be having an autoimmune hemolytic anemia (not due to blood transfusion).
General RBC transfusion rules that the blood bank follows:
ABO:
- Patients with Group O blood are considered universal donors (they can donate blood to all ABO blood types).
- Patients with Group AB blood are considered universal blood recipients (they can get blood from all ABO blood types).
RhD:
- We almost always give Rh-D negative blood to Rh-D negative patients. Note, the blood bank uses the “d” designation as another way of saying D-negative. In other words, Rh-D negative is the same as Rh-d. Even better, Rh-(d) vs Rh+(D) is accepted nomenclature.
- Under exceptional circumstances, Rh+(D) blood will be provided to Rh-(d) patients.
Emergency situations in a patient with unknown blood type:
- Cis-gender women and Trans-gender men of childbearing age are given O, Rh negative blood (ie Rh-(d)).
- Cis-gender men are given O, Rh positive blood. This is because there is way more O, Rh positive blood in the donor pool and cis-gender males can’t get pregnant, so we don’t worry about hemolytic disease of the fetus/newborn in a future pregnancy in cis-gender males.
- Pediatric patients: Institutional policies vary. Presumably cis-gender females would be given O, Rh negative blood as above.
We are now going to talk about some special clinical scenarios involving transfusion. We’ll then go on to discuss risks of transfusions.
Trauma Settings and Need for Massive Transfusion:
In settings of significant trauma with massive hemorrhage/blood loss (think severe car accidents, other types of trauma) the patient experiences hypoperfusion due to the blood loss. Hypoperfusion results in several problems, including hypothermia (due to lack of perfusion), endothelial activation (due to trauma), endogenous anticoagulation (including a rise in activated protein C), increased reactive oxygen species, and acidosis. All these problems contribute to an endogenous coagulopathy in the form of worsening bleeding tendency. These problems lead to platelet dysfunction, inactivation of coagulation proteins (and consumption of them as well), and hyperfibrinolysis. At the same time, often patients in massive trauma situations are given IV fluids as a first line of resuscitation, which further dilutes clotting factors and platelets (iatrogenic coagulopathy). Patients on blood thinners and antiplatelets also have an iatrogenic bleeding tendency, for obvious reasons.
(Note, if you have questions on why trauma can result in endogenous anticoagulation, an excellent paper to read is Cohen et. al., “Critical role of Activated Protein C in Early Coagulopathy and Later Organ Failure, Infection, and Death in Trauma Patients”, Ann Surg. 2012 Feb 255(2), 379-385. However, this detailed knowledge is beyond the scope of this course and you aren’t expected to know the material in this paper.)
Hence, in the setting of severe hemorrhage, patients not only need to be given PRBCs, but they also need to be given platelets and plasma/clotting factors as well. With this in mind, we have rules about what to give in what ratios, and this is the Massive Transfusion Protocol. The slide below depicts this protocol nicely.

Transfusion in the setting of Liver disease
The liver produces both pro-coagulant (clotting factors) and anticoagulant (protein C and S and others) proteins. In patients with significant liver disease, they will frequently have abnormal coagulation results. They often have a prolonged INR due to lower clotting factors and also often have low fibrinogen. However, proteins C and S are also often low. Patients with liver disease may also have a low-grade chronic DIC. Thus, in general for patients with liver disease, their tendency to bleed is balanced by their tendency to clot. Hence, patients with liver disease who aren’t bleeding don’t need prophylactic transfusions of the various blood products, despite the anemia, low platelets, and lower clotting/anticlotting factors that they typically have. Like patients without liver disease, they are generally transfused for clinical symptoms. (The main issue with patients with liver disease is to avoid invasive procedures unless they are absolutely necessary, as procedures can tip the scales towards bleeding, etc.).
Transfusion in patients with Sickle Cell Disease
We have previously discussed the diagnosis of sickle cell disease. These patients have 2 abnormal beta-globin genes with a specific mutation that ultimately leads to the production of hemoglobin S. Patients with RBCs with mainly hemoglobin S develop sickling of those red blood cells in low oxygen settings. This results in chronic hemolysis. These patients have chronic anemia due to chronic hemolysis of sickled RBCs. One of the mainstays of treatment for patients with Sickle Cell disease is blood transfusion. These patients are frequently/chronically transfused, which puts them at significant risk for alloimmunization against different RBC antigens and thus at risk for hemolytic reactions to transfusion. To prevent alloimmunization, many blood centers provide antigen matched RBC Units for more antigens than just ABO/Rh. The units are prophylactically matched for Rh (D, C, c, E, e) and K antigens. Sickle cell patients are at higher risk for delayed transfusion reactions (this occurs when they develop an antibody that reacts to the transfused blood later on, after the patient’s body has revved up the antibody production in response to another alloantigen). Hyperhemolysis syndrome, in which the patient hemolyzes both transfused RBCS AND their own RBCs, is a rare and life-threatening complication of transfusion in Sickle cell disease.
Indications for Transfusion in patients with Sickle cell disease are as follows:
- Severe anemia causing symptoms.
- Stroke—typically in this setting exchange transfusion is done.
- Pregnancy
- Surgery
- Acute vaso-occlusive episodes with pain crisis. Depending on the situation, exchange transfusions, simple transfusions, or no transfusions may be needed. However, most hematologists opt for exchange transfusions if transfusions are needed in this situation.
Lack of Response to transfusion:
RBCs:
It is rare to have a lack of response to RBC transfusion. If RBCs are crossmatched properly and the patient doesn’t react to them, they will improve oxygenation, etc. Recall that 1 unit of PRBCs generally raises the hgb in a patient by 1 g/dL. However, if the same patient is being hydrated at the same time, the rise in hgb may not be seen. Also, there is some lab variability/range of error that can affect the hgb result. Generally, a lack of response to RBCs means that the patient is still bleeding or is due to hemodilution. Lastly, if the patient is having a lot of blood draws, the lack of response to a transfusion of 1 unit of PRBCs may be partly iatrogenic.
Platelets:
It is not uncommon for patients who have had a lot of platelet transfusions to become refractory to platelets. In this setting, the platelet count doesn’t improve despite platelet transfusions. Platelet refractoriness is defined as a lack of adequate response to platelet transfusion. We generally expect the platelet count to rise by 30,000 to 50,000/uL after 1 apheresis unit (single donor unit) of platelets is administered. Informally, if we don’t see appropriate response to platelet transfusions, we become concerned that the patient is refractory to platelets.
You do not need to know about CCI/Corrected Count Increment, but this is the formal method used to determine if a patient is refractory to platelet transfusions. This is calculated as follows (do not memorize this):
(Platelet count after platelet transfusion – Platelet count before transfusion) X Body surface area
Platelet count in the bag
Generally, within 1 hour after platelet transfusion the CCI should calculate to 5000 to 7500/uL.
If the CCI on 2 separate occasions is < 5000/uL, then the patient is considered refractory to platelet transfusion.
Causes of Platelet refractoriness:
- Non-immunologic causes: Splenomegaly, Fever, Infection, Bleeding (this is presumed to cause platelets to be used up as well as bled out).
- Immunologic: Antibodies that bind platelets. (These can be antibodies to HLA, also called class I antibodies, or can be anti-platelet antibodies).
How do we test for platelet antibodies as a cause for platelet refractoriness due to Antibodies?
Platelets are known to express the following antigens:
- ABO
- Platelet antigens (HPA)
- HLA class I antigens
Platelet transfusions can result in HLA alloimmunization. In this setting, the recipient of platelets develops antibodies to certain HLA antigens and then if the next donor platelets have any of these immunogenic HLA antigens (the next time the patient is transfused), the patient’s antibodies bind the immunogenic HLA antigens and the body removes these Ab-bound platelets from circulation. This can be tested for using something called a PRA test (Panel Reactive Antibody test). If this is positive for HLA antibodies, then the patient will require HLA-matched (HLA-antigen matched) platelets for future platelet transfusions.
Risks of Transfusion:
The main risks of blood product transfusion are the following:
- Transfusion transmitted diseases: There are many potential diseases that can be transmitted by blood transfusion.
- Transfusion reactions
- Sepsis
- Transfusion Related Acute Lung Injury (TRALI)
- Transfusion Associated GVHD (this is almost uniformly fatal)
We’ll now discuss these main categories:
Transfusion Transmitted diseases: These include bacterial infections, viral infections, prion-associated diseases, fungal infections, etc. We work to mitigate transmission of infection by prevention.
Prevention of Transfusion transmitted diseases. Over the past 40 to 50 years, we have dramatically reduced the risk of infections due to transfusion. This is largely due to the following, which are the hallmarks of a safe blood program:
- Self-deferral: Self-deferral refers to the fact that we only take blood from volunteers. People are NOT paid to donate blood. There is no incentive to donate blood other than a desire to help. 100% of USA donations are due to people volunteering to be blood donors. (This is not the case for plasma donors, but that is used in a different setting and is processed such that infections are not likely.) Due to part of this process, we actively discourage family/friends from asking specific family members or friends to donate to them directly, as this may incentivize such people to donate when they actually shouldn’t be donating and such family members/friends may, based upon this pressure, fail to disclose behaviors that would exclude them from being donors. However, family/friends can be recruited to donate blood to help others, which is called replacement blood donation (they donate to replace the blood supply that the patient has utilized from the blood bank).
- Donor questionnaire: This is required for every donor to fill out. This questionnaire is used to identify risk factors for infectious disease that would then exclude the person from being a blood donor.
- Laboratory testing: Once a person volunteers to be a donor and is not ruled out as a potential donor by the donor questionnaire, they then donate blood. Once this blood is donated, it is tested for a variety of infectious agents.
Of note, 100% of USA donations are from volunteers. However, in other countries around the world there are not enough volunteer donations to cover the population’s transfusion needs.
With regards to the donor questionnaire, it is looking for whether the donor has risks for diseases we cannot test for and whether the donor has high risk behaviors putting them at risk for diseases that we can test for but may not be detected in the early stages of infection.
- Disease we cannot test for: Classical Jacob-Creutzfeldt, variant Jacob-Creutzfeldt, Malaria, other diseases we don’t know about.
- Disease we test for in blood donors: HIV, Hepatitis B, Hepatitis C, Syphilis, Chagas, West Nile Virus, HTLV-I and HTLV-II (HTLV stands for human T-cell leukemia virus), and bacterial infections/sepsis.
Risk of Contracting Disease from Blood Transfusion

We generally use Filtration to reduce risks for CMV, etc.
We can also irradiate blood products, which significantly reduces the risk for transfusion-related GVHD, but doesn’t decrease risk for infection.
Other methods of reducing risks of infection from blood products include the following: Photoactive component exposure, Illumination, Amotosalen + UV light (Intercept Technology) for platelets only. While these other methods are effective in decreasing residual risks of infection (which are already very small) and can replace irradiation to prevent GVHD, these are expensive and there is a relative loss of efficacy of the product. In general practice, these additional methods aren’t used.
Transfusion Reactions:
Transfusion reactions can refer to any patient reaction to blood product transfusion. Per Suddock et al, in their book, Transfusion Reactions (found on PubMed.gov), Transfusion reactions are defined as adverse reactions associated with transfusion of blood products. Reactions can occur during the transfusion (acute transfusion reactions) or days to weeks later (delayed transfusion reactions). A reaction may be immunologic or non-immunologic.
Transfusion reactions are generally classified as follows:
- Acute Hemolytic Transfusion reaction.
- Delayed Hemolytic Transfusion reaction.
- Febrile non-hemolytic transfusion reaction.
- Anaphylactic Transfusion reaction.
- Simple allergic transfusion reaction.
- Septic (bacterial contamination) transfusion reaction.
- Transfusion-related Acute Lung Injury (TRALI)
- Transfusion-associated circulatory overload (TACO).
In a patient who is has a reaction during a transfusion, it is important to do the following:
- STOP the transfusion. This is critical.
- Keep the IV line open (don’t pull the IV). We generally start an IV drip to keep the line open.
- Perform a clerical check of the product and the patient to ensure that the problem isn’t a mix up on product for patients in the hospital (this should have been avoided with the transfusion precheck process that the RNs do before they administer blood, but important to double check again if the patient has developed a reaction).
- Monitor Signs and Symptoms of the patient and treat with supportive measures as needed.
- Follow and document Vital signs.
- Give supportive treatments such as antihistamines for rash, medications to suppress fever caused by transfusion, medications for pain/rigors, etc.
- Send lab tests from the patient depending upon the clinical scenario.
- Note the type of product that is being transfused (platelets, RBCs, etc)
- Notify the blood bank and send a patient tube (blood draw from patient) AND the product causing the reaction to the blood bank for further testing.

Risk of Death from Transfusion:
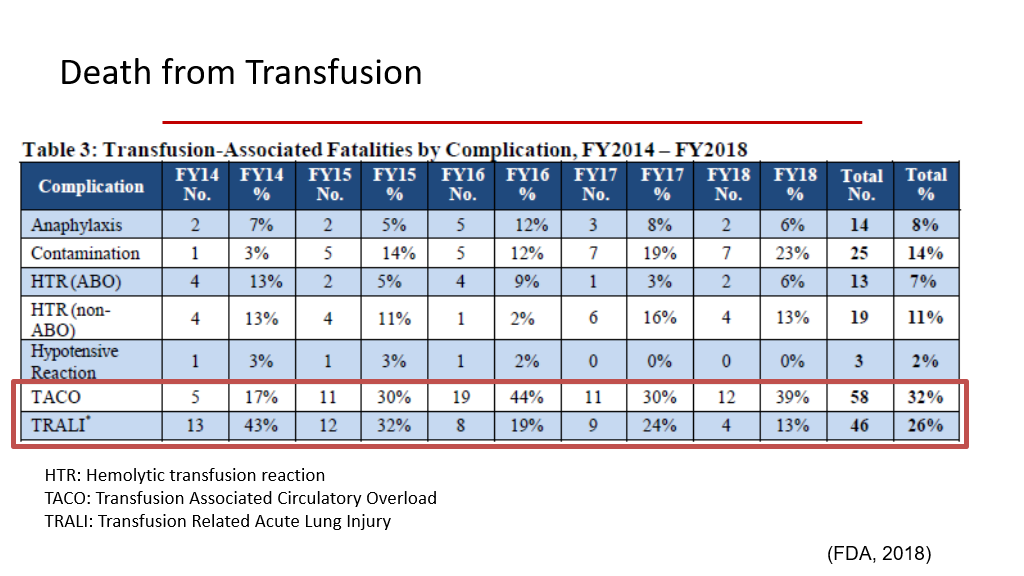
Risk of Death from Transfusion is rare, but risk of death is highest from TRALI and TACO. We’ll learn more about those types of transfusion reactions a little later in this course pack.
Acute Hemolytic Transfusion Reaction
This reaction is hemolysis due to the administration of antigenically incompatible red cells that occurs acutely with transfusion. By far the most common cause, if not the only cause, is error. The patient’s clinical presentation depends on how much blood was infusion before the error was suspected and the transfusion was stopped. Patients generally develop fever/chills which is often accompanied by nausea/vomiting, dyspnea, tachycardia, hemoglobinuria and severe pain (pain is usually in the lower back). Transfusion of a large volume of ABO-incompatible blood often produces hypotensive shock, oliguria, and DIC. Note that these symptoms may easily be missed in patients who are unconscious or under anesthesia (in the operating room, etc.). Laboratory testing will show Hemoglobinemia, Hemoglobinuria, elevated LDH, and a positive direct antiglobulin testing (positive Coombs test). Patients generally have active intravascular hemolysis due to IgM antibody or due to complement-fixing IgG. These days acute hemolytic transfusion reactions are rare due to rigorous blood testing by blood banks for crossmatching as well as pretransfusion checking processes by RNs/blood bank/etc. to ensure that the right product is going to the right patient.
Delayed Transfusion reaction
Delayed transfusion reactions are more common and are seldom due to error. In the case of delayed transfusion reactions, less immunogenic antigens on the transfused product trigger the blood recipient’s immune system and antibodies are produced, usually an amnestic response to a more remote trigger, leading to a delayed transfusion reaction. For example, think of a woman who was previously pregnant and exposed to C, inducing an anti-C antibody, but this was remote and she no longer expresses anti-C antibodies in a titer high enough to detect with a crossmatch. Because of this, when she is later crossmatched for blood needed at another time in her life, the cross match doesn’t pick up any anti-C antibody. she is given blood with C antigen present, and this triggers an amnestic antibody response—this generally occurs about 3 to 10 days after the exposure/blood transfusion. These antibodies then bind to the transfused RBCs and are taken out of circulation. In this setting, the hemolysis is usually extravascular and is not as severe as that seen in an acute reaction. With a delayed transfusion reaction, patients can present with a hemolytic anemia with low Hgb, high bilirubin, high LDH, spherocytosis, reticulocytosis, positive antibody screen (positive indirect antiglobulin testing or indirect Coombs test), and Positive DAT (direct antiglobulin testing)/positive Direct Coombs test.
TABLE 25-2Hemolytic Transfusion Reactions
|
|
Acute |
Delayed |
|
Timing |
Immediate |
3-10 days after transfusion |
|
Mechanism |
Preformed antibody |
Anamnestic antibody response |
|
Antibody |
IgM or complement-fixing IgG (e.g., anti-A or anti-B) |
IgG, no complement fixation (e.g., anti-Rh) |
|
Site of hemolysis |
Usually intravascular |
Usually extravascular |
|
Clinical sequelae |
Severe cases: shock, disseminated intravascular coagulation, acute renal failure |
Usually none |
|
|
|
|
Febrile (Non-hemolytic) Transfusion Reaction
This is a relatively common transfusion reaction. It is generally caused by cytokine release from donor WBCs that are present in the sample. The risk of this type of reaction has been reduced by leukoreduction/filtering of blood products, which removes most of the WBCs (leukoreduction reduces risk of febrile transfusion reaction from 2% to 1%). Signs/symptoms of a febrile (non-hemolytic) transfusion reaction are fever/chills within a few hours of transfusion. Nausea/vomiting/hypotension can also be seen. When a patient has this type of reaction during a transfusion, the transfusion should be stopped. We then support and monitor the patient and do the work up for a transfusion reaction, as above. However, if no hemolysis is found and no cause for ABO mismatch, etc. is found, then it is okay to proceed with transfusion of the next (another) product.
Urticarial/Allergic reaction to transfusion
Generally the symptom is that of a patient developing hives during a transfusion. This is due to donor proteins causing an IgE-mediated histamine release in a patient. Patients generally have symptoms of flushing, itching, or hives. The patient usually doesn’t have a fever. Treatment is to give antihistamines (and of course stop the transfusion first). If the patient’s symptoms subside/stabilize with antihistamines and patient is stable without other concerning symptoms/findings, it is generally ok to res-start the transfusion, but administer as a slower infusion.
Anaphylactic Transfusion reaction
Patients with an anaphylactic transfusion reaction present during a transfusion with hypotension, dyspnea, airway edema, anxiety, and larger or even full body rash. This requires emergent care. In addition to stopping the transfusion, patients are given antihistamines. Epinephrine, corticosteroids, pressors, and intubation may be necessary. In such a reaction, we generally wash future RBC or platelet blood products for this patient.
Sepsis
Sepsis with a transfusion is due to blood components contaminated by bacteria. The most common causes are bacteria from blood donor skin flora or unrecognized blood donor bacteremia. Occasionally environmental or product handling during processing can result in bacterial contamination. Always let the blood bank know if a septic reaction is suspected, as blood products should then be cultured to identify bacteria, etc.
Transfusion Related Acute Lung Injury (TRALI)
TRALI is an uncommon but potentially deadly transfusion reaction. About 1/10,000 patients develops acute interstitial pneumonitis withing 6 hours of transfusion, AKA TRALI. Symptoms include dyspnea, tachycardia, hypoxia, fever, and hypotension. CXR in this setting will show rapid development of bilateral pulmonary infiltrates. It can be very difficult to distinguish TRALI from cardiogenic shock/cardiogenic pulmonary edema, pulmonary hemorrhage, or acute respiratory distress syndrome (ARDS). Blood donor antibodies that attack the patient’s pulmonary endothelial cells or bind to and activate the patient’s neutrophils are thought to be the cause of the problem. Frequently, the antibodies appear to be against HLA antigens. Other cases are associated with antibodies against a polymorphic antigen expressed on neutrophils, called choline transporter-like protein (CLT2). The treatment of TRALI is supportive: oxygen is administered, and intubation is frequently needed. Risk of TRALI can be reduced by use of plasma from cis-male donors (or from women or trans-men who have not yet been pregnant), as women/trans-men often become HLA-alloimmunized during pregnancy. Cases of suspected TRALI are investigated by the blood bank. Donors of blood products believed to have caused TRALI are PERMANENTLY banned from donating blood.
Transfusion-associated circulatory overload (TACO)
TACO is a problem caused by the transfusion contributing to excess fluid in the circulatory system that results in hypervolemia. Symptoms of TACO include shortness of breath, hypoxemia, leg swelling/peripheral edema, hypertension, and tachycardia that occur within 12 hours of a transfusion. It can be difficult to differentiate TACO from TRALI, but generally peripheral swelling and hypertension are more common with TACO. Risk factors for TACO include a large volume of blood products administered to the patient. TACO is more of a risk in patients who already are prone to fluid/volume overload—such as patients with liver disease, heart disease, and/or kidney failure. Management of TACO includes immediately stopping the transfusion (as with any other transfusion reaction), giving supplemental O2, and then giving diuretics to remove excess fluid.
Transfusion Associated Graft vs Host Disease (Transfusion Associated GVHD, TA-GVHD)
This is an almost uniformly fatal transfusion-related problem that results if Donor lymphocytes engraft and proliferate in a recipient of a blood product. Severely immunocompromised patients are at risk for this. Also, immunocompetent patients are at risk for this if they are transfused with HLA-haploidentical blood products (ie, blood products from a relative). GA-GVHD occurs when the blood product recipient has a tolerance for the donor lymphocytes, due to similar HLA make-up, so the recipient’s WBCs don’t see the rare donor lymphocytes in the blood product as foreign and thus don’t destroy them. This allows the donor lymphocytes to engraft in the bone marrow of the patient who received the blood transfusion. However, the Donor Lymphocytes may see the recipient’s lymphocytes/WBCs as foreign and destroy them. It is for this reason that patients are strongly discouraged from getting blood transfusions from family members. Once engrafted, the donor lymphocytes attack the patient’s own hematopoietic cells resulting in refractory cytopenias. These cells can also attack other systems and cause fever, enterocolitis, rash, hepatitis, etc. TA-GVHD is usually fatal—90% to 100% of patients who get this die from this.
Irradiation of blood products prevents TA-GVHD. Irradiation inactivated lymphocytes so that they can’t continue to divide. Hence, irradiated blood product lymphocytes can’t reproduce in the recipient.
Who should get irradiated blood products?
- Fetuses/Newborns
- Bone marrow transplant candidates and Bone marrow transplant recipients.
- Patients with a hematological malignancy (leukemias, lymphomas, etc.)
- Patients with congenital immunodeficiency (DiGeorge Syndrome, etc.)
Note, Pathogen-reduced platelet units (see prior discussion above re: pathogen inactivation Technology) already have caused the cells in the product to be unable to replicate, so these types of platelet units don’t also require irradiation. However, Pathogen-reduced platelet units are rarely done, so usually platelets are irradiated, as above.
Blood Product Transfusion Consent
Due to risks of transfusions, as above, it is important to obtain informed consent from patients for blood transfusion. Informed consent is obtained prior to blood product administration unless the patient is in a life-threatening situation and needs products and is unable to consent. As part of the blood transfusion consent process, the following is discussed:
- Indication: We explain to the patient why a transfusion is needed.
- Benefits: We discuss the benefits to the patient for the transfusion.
- Risks: We explain the risks of getting the blood product to the patient.
- Alternatives to transfusion: We discuss alternatives to transfusion, if such alternatives exist.
- Opportunity to ask Questions: We provide the patient, during the consenting process, the opportunity to ask questions about everything we’ve discussed and answer any other questions that they may have.
Authors and Contributors:
Corliss Newman, MD authored this course pack material based partly upon slide lecture material prepared by Rida Hasan, MD. Nicholas Burwick, MD and Kristi Rice, MD provided editing assistance. Additional sources of information utilized in preparation of this course pack included chapters from Pathophysiology of Blood Disorders by Aster and Bunn and Wikipedia.