
6 Clinical evaluation of Coagulation Defects
Student Learning Objectives:
- Describe the basis coagulation tests and how they related to the clotting cascade.
- Interpret the laboratory tests used to evaluate patients with suspected abnormalities in platelet function.
- Interpret laboratory tests used to evaluate patients with suspected abnormalities in coagulation protein function or amount.
- Recognize the differences in clinical and laboratory findings between a patient with a platelet function defect, a coagulation protein defect, and a defect in fibrinolysis.
- Recognize the clinical presentation and laboratory results of selected inherited or acquired coagulation disorders.
- Apply your knowledge of hemostasis to the clinical evaluation and management of a patient with a bleeding disorder.
USMLE Content Descriptors: Cytopenias, Hypocoagulable, Hypercoagulable, Congenital disorders.
Optional textbook reading: Part II Hemostasis and thrombosis, Chapter 16: Acquired Coagulation Disorders (section on Vitamin K deficiency and Liver disease)
Introduction:
In a prior session, we focused on general knowledge of clotting and then spent time discussing problems with primary hemostasis, secondary hemostasis, and additional platelet problems associated with low platelets and clotting. Here we move on to further discuss problems with bleeding and how to assess the cause of bleeding. We will particularly look at labs that assess the clotting cascade (secondary hemostasis). We’ll also discuss some disorders associated with increased blood clots.
Although complex, clotting and bleeding problems can be broken down for evaluation by the following:
- Is there too much bleeding?
- Is there too much clotting?
- Are there problems with both bleeding and clotting?
Causes for too much bleeding: Platelet dysfunction, Clotting Factor Deficiencies, Increased Fibrinolysis.
Causes for too much clotting: Deficiency of natural anticoagulant factors (Protein C, Protein S, and Antithrombin), Activated protein C resistance (Factor V leiden mutation), Too much of a clotting factor (excess VIII, excess Prothrombin), certain autoantibodies that promote clotting (certain antiphospholipid antibodies, aka “Lupus Inhibitor”).
Causes both bleeding and clotting: Disseminated Intravascular Coagulation (DIC).
Causes for too much bleeding:
In a patient who presents with a problem bleeding, it is important to figure out where the defect in hemostasis is that is leading to bleeding. Again, recall that the hemostasis involves the following:
Endothelium exposed due to vessel wall damage -> Primary Hemostasis (platelet plug) -> Secondary Hemostasis (clotting cascade) – > Fibrinolysis.
Hence, defects in any of the above can lead to problems with bleeding.
Endothelial causes of bleeding: A disorder of the collagen/vascular system can result in bleeding problems. This can be seen in Ehler’s Danlos or Marfan’s syndrome, which are both congenital. This can be acquired in patients with scurvy. These patients tend to have bruising/bleed more easily with tissue damage because their tissues are more fragile. There are no coagulation screening tests that can predict these problems, nor is there effective treatment. Endothelial causes of bleeding are a diagnosis of exclusion, generally.
Bleeding problems due to Primary Hemostasis problems: As we discussed in the last lecture series, a disorder in platelet quantity or quality/function results in bleeding. Also, a quantitative or qualitative defect in von Willebrand factor results in bleeding problems. This bleeding is generally mucocutaneous: easy bruising, epistaxis, oral bleeding, GI bleeding, and menorrhagia. Clotting tests that diagnose problems with Primary Hemostasis involve platelet function testing, a more detailed platelet aggregation study, and tests to detect problems with Von Willebrand factor. There are effective treatments for disorders of primary hemostasis.
Bleeding problems due to Secondary Hemostasis problems: As discussed previously, secondary hemostasis involves the clotting cascade and results in a more permanent clot by interaction of various clotting factors to produce crosslinked fibrin. Defects in clotting factors can result in bleeding in deep tissue or joints, although other types of bleeding can be seen as well. Hemophilia patients, for example, were very prone to joint defects due to recurrent joint bleeds prior to the onset of therapies to prevent bleeding in such patients. Defects in the clotting cascade can be detected with screening lab tests PTT (partial thromboplastin time), PT (prothrombin time), and TT (thrombin time). Specific factor activity level assays can then be utilized to further narrow down the problem at hand. There are effective treatments for disorders of secondary hemostasis.
Fibrinolysis: After formation of a fibrin clot, fibrinolytic enzymes are responsible to break down the clot after endothelial regeneration has occurred. Overactive fibrinolysis will result in an unstable clot and increased bleeding. In such patients, bleeding is usually delayed, occurring after clot formation. A typical story is that patients initially clot normally, but then have bleeding occur unexpectedly a while later. Disorders of fibrinolysis are less common than disorders of primary or secondary hemostasis. There are effective treatments for disorders of fibrinolysis resulting in bleeding.
In approaching cases of bleeding in the clinical setting, it can be useful to consider defects in either primary hemostasis (platelet plug) or secondary hemostasis (coagulation factor hemostasis) based upon the type of bleeding, all though there can be some cross over of types of bleeding. For example, in a patient with mucocutaneous bleeding, we tend to start our work up focusing more on evaluating primary hemostasis and quickly ruling out secondary hemostasis problems. In a patient with deep tissue and joint bleeding, we would be more focused on a secondary hemostasis problem. Thus, knowing the patient’s bleeding pattern is useful to guide the laboratory work up.
So, in evaluating patients with a bleeding problem, the patient’s history is very important. Is there a personal or family history of a collagen vascular disorder? What type of bleeding is the patient having—where is it, when does it occur, etc.? It is particularly important to ask females about menses. A woman with a family h/o heavy menses, for example, may think it is normal to need to use a rubber sheet to prevent blood staining of a mattress during her usual heavy period, because her mother did this too! This can be a sign, of a family h/o heavy bleeding that the patient wouldn’t necessarily think is abnormal (even though it is) and wouldn’t think to mention to you!
Evaluating the patient with a bleeding disorder: There are 2 steps.
- Take a bleeding history.
- Proceed to laboratory testing.
Take a bleeding history: This is critical.
Personal bleeding history: Find out about childhood bleeding/bruising, bleeding with dental work or dental extractions, frequency/severity of nose bleeds, age of menarche and menstrual history, surgical history, history of anemia/iron deficiency/blood transfusions.
Find out about medications or supplements that the patient is taking.
Be aware of other past medical history (be on the lookout for medical problems that may indicate a collagen vascular disorder).
Review family history for family members who may have had bleeding problems.
Laboratory testing: Typically, we start with checking a CBC, which includes a platelet count, as well as a PTT, PT, and sometimes TT.
The PTT, PT, and TT are sometimes called coagulation screening. We have previously discussed the CBC and platelet count. Let’s spend some more time talking about the PTT, PT, and TT (Partial Thromboplastin Time, Prothrombin Time, and Thrombin Time).
The PTT, PT, and TT are the three basic labs that measure the clotting cascade (secondary hemostasis). The following slide illustrates what they measure very nicely. (Note that a more specific test of the PTT is the aPPT or activated Partial Thromboplastin Time. Regardless of PTT or aPPT nomenclature, we are talking about basically the same test here—see below to know the subtle difference between aPTT and PTT.) Recall that the coagulation cascade is measured this way as it occurs in a test tube, which is slightly different than what occurs in vivo (we covered this in the previous lecture).
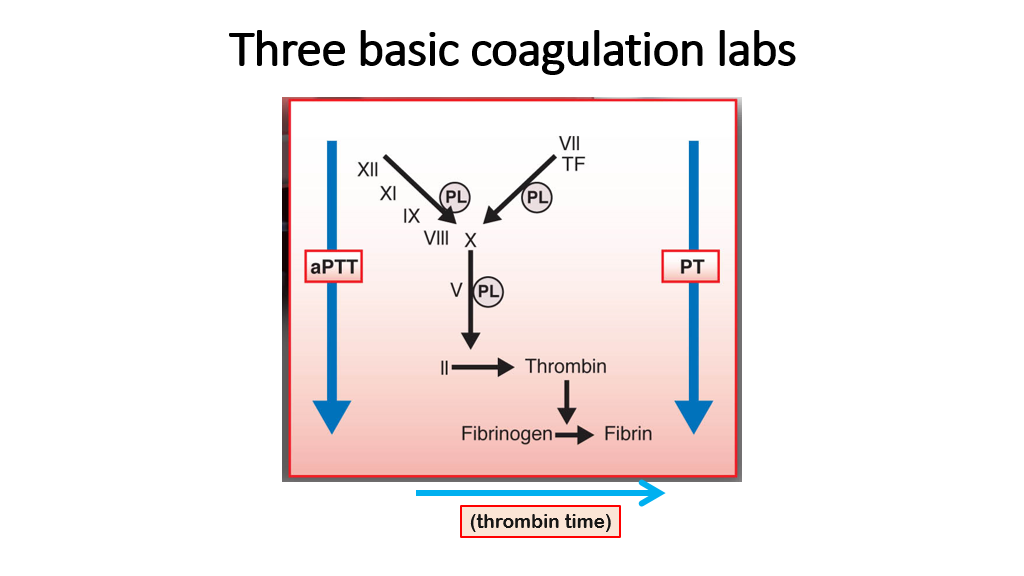
We’ll now discuss these three coagulation labs in detail:
PT
The Prothrombin Time, aka PT, measures activity of factors VII, X, II (recall that Thrombin is activated factor II), and conversion of Fibrinogen to Fibrin. To perform a PT, the lab measures the time it takes to form a clot when tissue thromboplastin and calcium are added to plasma. Tissue thromboplastin, FYI, is an extract of brain containing large amounts of phospholipid and tissue factor. The prothrombin Time is reported in seconds, and is also reported as an INR, to allow comparisons across labs.
The PT is very sensitive to levels of factor VII. The PT will be affected by deficiencies in VII, X, V, Thrombin, and Fibrinogen. Since factor VII is a vitamin K-dependent factor, the PT is a good way to assess levels of vitamin K-dependent factors, which are decreased with warfarin therapy. The PT will be prolonged with significant Vitamin K deficiency. Also, in the setting of liver disease, vitamin K-dependent gamma-carboxylation of factors II, VII, IX and X (as well as protein C and protein S) is decreased. Hence, the PT can be prolonged in liver disease severe enough to cause deficiencies of these factors. Inhibitors of factors X and II (VII inhibitors and Fibrinogen inhibitors are rare/unheard of) can also prolong the PT.
The INR: We discussed this in detail as an aside above. However, to review (this is important, as you’ll see INRs for your entire medicine career), the INR (international Normalized Ratio) was devised to allow comparison of prothrombin times between laboratories, as the actual PT will vary depending upon the lot/manufacturer of the tissue thromboplastin that is used by the lab in doing the PT assay.
The INR was initially intended for use to determine/standardize the proper dosing of warfarin across different labs/different tissue thromboplastin reagents because the PT can vary depending on the source of tissue thromboplastin used in the assay. The INR is based on an experimentally determined factor for a given preparation of tissue thromboplastin which is called the international sensitivity index. Each manufacturer of tissue thromboplastin will also give the ISI of the tissue thromboplastin that they produce. The gold standard is human brain thromboplastin, which has an ISI of 1. Basically, the ISI allows the ratio of the patient PT to the geometric mean of a normal population PT to be adjusted based on the laboratory performance of a given preparation of tissue thromboplastin that is added to the tube to start the clotting reaction.
The INR is calculated as shown below:

aPTT
The Activated Partial Thromboplastin time (aPPT) is similar to the PTT, also called the Partial Thromboplastin Time: This test (aPTT or PTT) measures the following factors: XII, XI, IX, VIII, X, V, II, Fibrinogen as they work to form a fibrin clot. In the slide above showing the 3 clotting tests, recall that another name for IIa is Thrombin. Other factors that are measured in this intrinsic clotting pathway that aren’t shown on the above slide are prekallikrein and high molecular weight kininogen (HMWK). Deficiencies of these factors will prolong the PTT. Other factors that cause a prolonged PTT include anticoagulants such as heparin, direct thrombin inhibitors, direct factor Xa inhibitors, and warfarin to a lesser extent. Liver disease can cause deficiencies in all factor levels except VIII, so liver disease can cause a prolonged PTT. Lupus inhibitors (certain antiphospholipid antibodies that interfere with the PTT assay) and specific factor inhibitors (example factor V inhibitor, factor VIII inhibitor, factor IX inhibitor) will also cause a prolonged PTT.
The aPTT is assayed by first incubating plasma with a reagent to activate the contact factors. (The contact factors are XII, HMWK, and prekallikrein). The activator speeds up the test/clotting time and allows us to get a narrower reference range. Calcium and phospholipids are then added to initiate the coagulation cascade.
The PTT is done the same way as the aPTT, EXCEPT that the activator is not added to the test first. Hence, the clotting time takes a little longer without the activator, but the same clotting pathway is still activated by contact with the glass walls of the test tube.
The PTT is reportedly less sensitive to heparin therapy than aPTT. Hence, most hospital labs use the aPTT, as this test not only measures clotting, but can be used to measure the degree of anticoagulation more correctly (compared to the PTT) when a patient is on heparin therapy. Heparin forms a complex with antithrombin that inhibits the activity of all the serine proteases of the intrinsic pathway (factors XII, XI, IX, IX and II). Heparin will cause a prolonged aPTT and a prolonged PTT. However, the aPTT is preferred to measure heparin therapy effectiveness (as in how prolonged the aPTT is) compared to the PTT. This knowledge about aPTT vs PTT is beyond the scope knowledge for this course, but questions frequently come up about the difference between PTT and aPTT. For all intents and purposes, hematologists use these tests interchangeably.
One last little pearl of wisdom: In an acute phase response (often inflammation, stress, etc), factor VIII and fibrinogen are increased. Can you predict what would happen to the aPTT in this situation? If you guessed that the aPTT may be shortened, you are correct!!
TT
The thrombin time (TT) reflects the conversion of fibrinogen to fibrin. This is done by adding human or bovine thrombin to platelet poor plasma at 37 C and measuring the time to formation of fibrin. Note that this Thrombin time only reflects fibrinogen, as the test is not measuring factor II in the patient. Instead, thrombin (activated factor II) is added as a reagent for the assay.
Factors that prolong the TT include the following:
- Anticoagulants: Unfractionated heparin prolongs the TT, although low molecular weight heparins do not. Direct thrombin inhibitors can prolong the TT. The TT is not recommended for monitoring of these agents. The TT is not prolonged by warfarin, as the test is only measuring the patient’s fibrinogen and not any of the vitamin K dependent clotting factors.
- Congenital: This is usually due to a deficiency of fibrinogen quantity or qualitative defects in fibrinogen.
- Acquired deficiencies of fibrinogen or acquired factors that prolong the TT: DIC will promote increased fibrin degradation productions, which, in turn, impair fibrin polymerization and lead to a TT. Amyloidosis and other elevated paraproteins interfere with conversion from fibrinogen to fibrin and thus prolong the TT. Liver disease will result in decreased production of fibrinogen. Thrombolysis breaks down clots and leads to a prolonged TT.
Putting it all Together for these 3 tests of the clotting cascade:
Let’s think of how we would interpret the following clotting findings in a patient with a bleeding problem and a normal platelet count leading us to look next at the PT, aPTT, and TT:
PT, aPTT, TT all normal: In a patient with bleeding, this generally rules out clotting cascade problems. In this case, one would go back and look for problems with primary hemostasis, a factor XIII deficiency, or problems with fibrinolysis. (Also, don’t forget to think about a tissue disorder such as a connective tissue disorder.)
PT, aPTT and TT all prolonged: This can be seen with consumption of all clotting factors—which is what occurs in DIC. This can also be seen with a primary problem involving fibrinogen/severely low fibrinogen. Massive doses of heparin can also do this. (Heparin doesn’t usually affect the PT, but it can if the levels are high enough).
aPTT/TT prolonged, PT Normal: We know it isn’t a factor VII problem! Since the TT is abnormal, the problem has to be with fibrin or a heparin anticoagulant or a direct thrombin inhibitor (usually a drug). Fibrin problems are rare. Most common cause is heparin contamination.
PT/aPTT prolonged/TT normal: It isn’t a problem with fibrinogen or an anticoagulant that affects the TT. Given that both PT and aPTT are prolonged, the problem must be something involved in the common pathway, so be looking at factors X, V, II.
PT/TT prolonged/aPTT normal: That’s weird and I’ve never seen this. If you can think of a scenario where this occurs, email Corliss Newman, MD, and she’ll be very impressed!
PT prolonged, aPTT and TT normal: It’s a factor VII problem, usually due to warfarin (warfarin tends to affect the PT, is unlikely to affect the aPTT, and doesn’t affect the TT).
aPTT prolonged, PT/TT normal: It’s got to be a problem with XII, XI, IX, or VIII.
TT prolonged, aPTT/PT normal: It’s a problem with fibrinogen generally. It can’t be an anticoagulant (heparin, direct thrombin inhibitor, etc, as these should also affect the aPTT.
Hence, you can see how knowledge of these 3 assays and the clotting cascade allows us to use these tests to figure out the clotting cascade problem, if indeed there is one. Also, as you can see, this ties information corresponds some to use of anticoagulants and how they work. We’ll have more on this later!
So, what do you do if a PT, aPTT, or TT is prolonged and the patient is not known to be on a blood thinner? First, you look at the pattern of the PT/PTT/TT results to see if you can figure out things, as above.
What if the PTT or aPTT is prolonged and the TT/PT are normal?
Answer: You do a 1:1 mixing study to determine if the prolonged PTT/ aPTT is due to a factor deficiency or due to an inhibitor of a clotting factor.
What the heck is a mixing study? Ah, that brings us to our next lab test discussion!
1:1 mixing study
This is generally done when an aPTT is prolonged (although, it can also be done to assess a prolonged PT or TT). In a mixing study, the patient’s plasma is mixed 1:1 with normal plasma and then the aPTT test (or PT or TT) is repeated. Since 50% factor is enough to achieve a normal PTT in most instances, factor deficiencies should correct upon mixing with normal plasma. For example, even if somebody has 0% factor VIII, then mixing 1:1 with normal donor plasma, would be sufficient to normalize the PTT. If you do a mixing study and the aPTT corrects to normal, then the problem that caused the originally prolonged aPTT is a factor deficiency.
If you do a mixing study and it doesn’t fully correct the aPTT to normal, then you are not dealing with a factor deficiency. Instead, the problem is due to an inhibitor that is present in the plasma and inhibits clotting factor function. A factor inhibitor in the patient’s plasma also will inhibit the factor in the normal plasma when it is mixed with the patient’s plasma, so a 1:1 mix of the patient’s plasma with normal plasma will still cause a prolonged aPTT result. Hence, when a mixing study is done and the aPTT doesn’t correct to normal, an inhibitor is expected to be the cause of the problem.
Question: What is the most common inhibitor causing a prolonged PTT or aPTT?
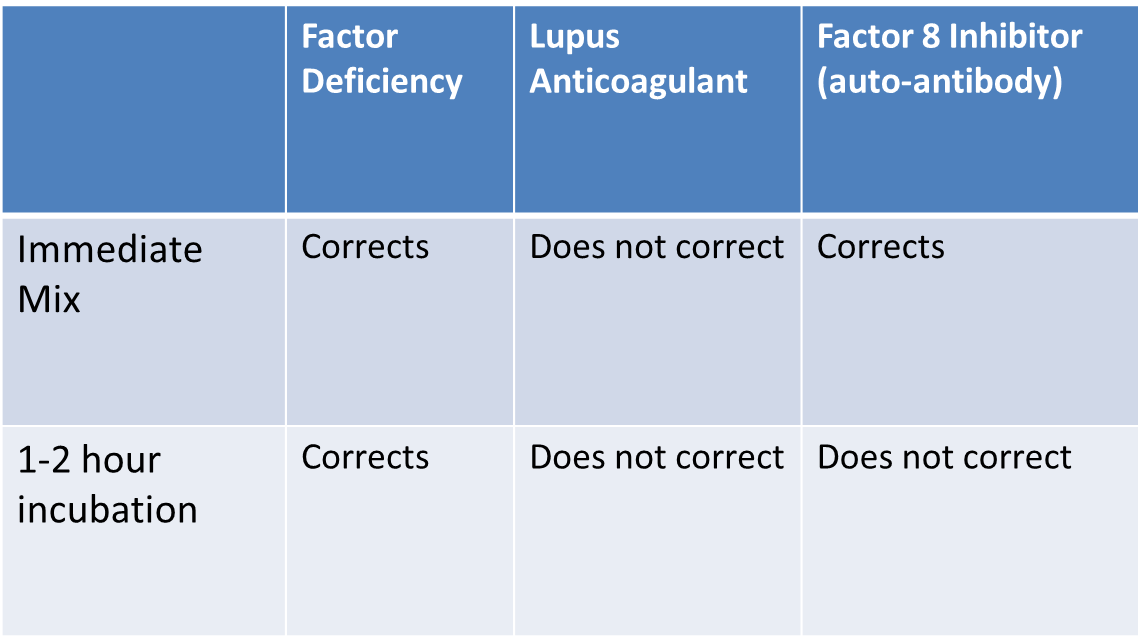
Answer: Lupus inhibitor. Lupus inhibitors are a notorious cause of a prolonged aPTT that is not actually clinically significant for bleeding. In fact, just the opposite, the presence of a lupus inhibitor is associated with increased risk of clotting (because lupus inhibitors are antiphospholipid antibodies—see the session on clotting disorders/risks). The elevation of the PTT by the lupus inhibitor is an in vitro artifact (the lupus inhibitor interferes with the assembly of the prothrombinase complex on phospholipids in the PTT assay).
Other causes of inhibitors: Acquired inhibitors to clotting factors can occur. Acquired (rather than inherited) Hemophilia A due to the development of auto-antibodies against factor VIII can also be a cause of severe bleeding, and in these patients, the aPTT doesn’t correct either (compare that to what happens in the inherited form). One subtle but important difference between a FVIII inhibitor (auto-antibody) and Lupus anticoagulant, is that with a FVIII inhibitor the 1:1 mix will initially correct, but then fail to correct after longer incubation (1-2 hours) due the slow reaction kinetics, whereas with a lupus inhibitor the 1:1 mix fails to normalize from the very start (because it impacts the assay itself).
Hemophilia:
Hemophilia is a bleeding disorder due to a clotting factor deficiency or inhibitor, which we previously discussed in the last lecture. The 2 main hemophilias (all hemophilias are relatively rare) are Hemophilia A and Hemophilia B.
Hemophilia A: this is an X-linked disorder where patients are deficient in Factor VIII. You can predict that such patients will have a bleeding history, and their work up will show a problem with secondary hemostasis. The 3 clotting assays in the setting of hemophilia A will show a normal TT and PT, but an abnormal aPTT that corrects with a mixing study. Further work up to assess for the specific clotting factors will show a lower factor VIII activity level. The treatment is to give recombinant factor VIII products.
Hemophilia B: This is an X-linked disorder where patients are deficient in Factor IX. You can predict that such patients will have a bleeding history, and their work up will show a problem with secondary hemostasis. The 3 clotting assays in the setting of hemophilia A will show a normal TT and PT, but an abnormal aPTT, that corrects with a mixing study. Further work up to assess for the specific clotting factors will show a lower factor IX activity level. The treatment is to give recombinant factor IX products.
More Laboratory testing: Primary Hemostasis:
We’ve talked about tests that measure the clotting cascade, which is the process of secondary hemostasis. Now, let’s talk about how we measure primary hemostasis. Recall that primary hemostasis involves the platelet plug and von Willebrand factor (VWF). Hence, obvious assays for primary hemostasis are going to assess platelets and VWF. Recall that in the process of primary hemostasis, collagen is exposed due to endothelial injury. Platelets then bind to collagen via the glycoprotein Ia/IIa receptor. VWF binds to collagen as well and then binds to platelets via the glycoprotein Ib receptor. In primary hemostasis, platelets also activate and release agonists (ADP, thromboxane, etc) to aggregate more platelets binding through the glycoprotein IIb/IIIa receptor. Any disorders in this process will lead to disorders of primary hemostasis. Lab tests that assess primary hemostasis include the following:
Measures of Platelet Function:
- Platelet Count (noted on the CBC).
- Platelet Function Assay (aka PFA-100).
- Platelet Aggregometry.
Measures of VWF: Von Willebrand Factor Assays: More below on how this is done.
Measures of Platelet Function:
We have previously covered a platelet count, which is measured in a CBC. Assuming a platelet count is normal, but a disorder of primary hemostasis is suspected, we would go on to measure platelet function and VWF in more detail as follows:
Note: It is important to clarify with a patient that they have not been taking antiplatelet drugs/NSAIDS, as antiplatelet drugs/NSAIDs will affect the platelet function assay…. So will SSRIs, so this can be challenging in a patient on SSRIs for depression…
PFA-100: Platelet Function Analyzer: The PFA-100 is a system in which citrated blood (the citrate is an anticoagulant) is aspirated at high shear rates through disposable cartridges containing an aperture withing a membrane coated with either collagen and epinephrine OR collagen and ADP. Collagen/EPI or Collagen/ADP are agonists that induce platelet activation and aggregation. As the platelets aggregate, they close the aperture so that blood flow through the aperture stops. This cessation of blood flow is called the closure time. The results are given as a time to closure with a normal range. A prolonged closure time indicates a platelet function problem, if one has ruled out low platelets, anemia or VW disease. A normal or shortened time is not considered abnormal. (A shortened closure time is not concerning—it is basically a super normal test.) The PFA or PFA-100 is affected by platelet count, so it is not useful in patients with a low platelet count. It is also affected by hematocrit (yes, severe anemia can prolong the PFA, presumably because the blood is less cellular), low VWF levels, and platelet function. Hence, to interpret a PFA result, one also needs to know a CBC result (to rule out anemia and low platelets) and a Von Willebrand Panel (more below, this rules out Von Willebrand disease).
If the PFA-100 is abnormal and we have ruled out anemia, thrombocytopenia, or VW disease, then the next step is to do a more detailed platelet function analysis in the form of platelet aggregometry.
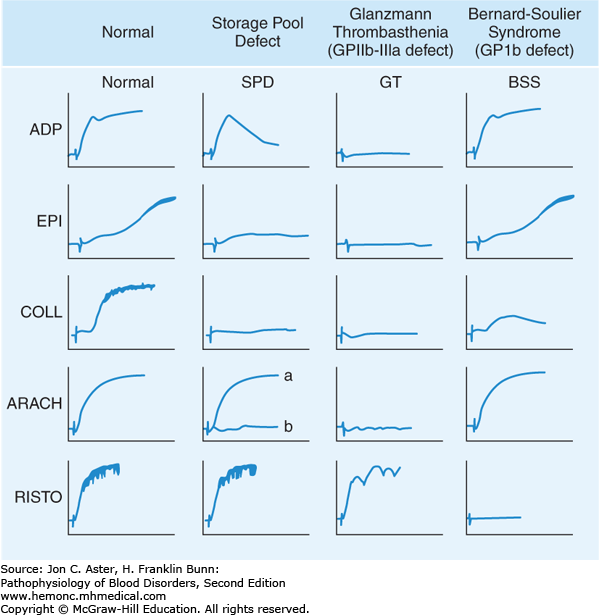
Platelet Aggregometry: In this testing, light is shined through a tube filled with platelet rich plasma. The platelets are dispersed/not activated, so the light passage is impaired somewhat by the dispersed platelets in the sample. An agonist is added (ADP, Arachidonic acid, epinephrine, collage, thromboxane, or ristocetin) and the platelets are activated and aggregate. As they aggregate/clump, increased light transmission occurs through the tube. The test measures patient platelet aggregation response to the different agonists and uses a control of a known normal platelet sample as a comparison test. The pattern of the patient’s platelet aggregation with different reagents can help determine what the underlying problem is. For example, in the case of Bernard-Soulier syndrome, which is a platelet problem where platelets have a mutation in the platelet receptor glycoprotein Ib, the platelets will aggregate normally with ADP, Epi, and collagen agonists, but not with Ristocetin, which acts via the glycoprotein Ib receptor. Patients with Glanzmann thrombasthenia have a bleeding syndrome due to the inheritance of a defect in the gene for either GPIIb or GPIIIa. As a result, their platelets lack docking sites for fibrinogen binding. Even though agonists such as adenosine diphosphate, arachidonic acid, epinephrine, and collagen can activate their platelets, aggregation fails to occur due to the absence of fibrinogen cross-linking. The only agonist that triggers cross-linking of Glanzmann platelets is ristocetin, which provides an alternative, non-physiologic mechanism for platelet aggregation. Patients with platelet storage pool defects have abnormalities either in the structure of the dense or alpha granules or in the ability to release granule contents during activation. Both types of defects lead to impairment of the “second wave” of aggregation that normally arises from the release of adenosine diphosphate from platelet granules. As a result, much higher doses of agonist are required for platelet activation.
Von Willebrand disease (VWD):
VWD is a hereditary disorder that causes decreased levels or function of Von Willebrand Factor (VWF). As you recall, VWF is involved in platelet aggregation for primary hemostasis. Hence, the other main cause of bleeding due to a primary hemostasis problem is VWD. VWD is the most common hereditary bleeding disorder, so it’s a good one to know. It is also important to know that there are various subtypes of VWD (did I mention previously that medicine is complicated?) and they have different inheritance patterns!
Also, although VWF is mainly involved in primary hemostasis, VWF also binds to factor VIII and stabilizes it. Hence, severe deficiencies of VWF can also result in low factor VIII levels, leading to problems with secondary hemostasis (did I mention previously that medicine is complicated?).
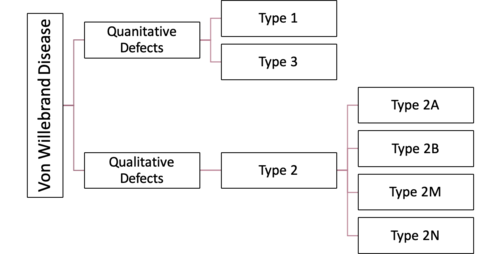
In broad terms, Von Willebrand Disease can be broken into 3 main types (there are additional sub-types that students do not need to know for this block). Type 1 and type 3 WVD are quantitative disorders of VWF. Type 2 disorders of VWD are qualitative disorders in the VWF that is produced.
Quantitative VWF defect (decreased VWF levels)
Type 1 VWD: This is by far the most common form of Von Willebrand disease, and accounts for about 75% of cases of VWD. This is inherited in an autosomal dominant fashion. Patients generally have mild bleeding symptoms. Patients frequently don’t require treatment unless they are going to undergo an invasive procedure or are in a severe accident, etc.
Type 3 VWD: This is very rare. Patients have absent VWF protein. Clinically, patients are similar to hemophilia A in their presentation (given that they do not have VWF to stabilize factor VIII, so VIII levels are also low) and will have joint bleeding in addition to mucocutaneous bleeding. Inheritance is Autosomal recessive.
Qualitative VWF Defect (Normal VWF levels, but decreased function/abnormal Function or variation in amounts of large multimers):
These are all called type 2 VWD, but they are further broken down into subtypes (2A, 2B, 2M, and 2N) due to the different qualitative defects that occur. You don’t need to know the various subtypes, but you should know that Type 2 VWD is generally a qualitative function problem, not a deficiency. The inheritance pattern of the different Type 2 VWD sub-types is variable and can be autosomal dominant or recessive.
(Graphic courtesy of Physiopedia)
How we test for Von Willebrand Disease:
To test for VWD requires ordering a Von Willebrand Panel. We test for the following:
- VWF antigen—this assesses the amount of Von Willebrand factor present in the blood.
- Platelet-dependent VWF activity—this assesses function of VWF.
- Collagen binding activity—this assesses function of VWF.
- Factor VIII level—this assesses function of VWF (recall that VWF stabilizes factor VIII).
- VWF multimers: This assesses the structure of VWF. Von Willebrand factor is present in the blood in varying sizes of multimers and we want to know that these levels are normal as well. For example, a certain subtype of VWD has normal VWF but decreased amount of large multimers.
Where is Von Willebrand Factor Produced and Stored, and how is it secreted?
VWF is synthesized in endothelial cells and in megakaryocytes (platelet precursor cells). It is stored in storage granules in endothelial cells called Weibel-Palade bodies and in alpha granules in platelets. It is secreted by endothelial cells into the subendothelial matrix and into circulation as varying sized multimers. Smaller VWF multimers are constitutively secreted from endothelial cells and megakaryocytes. Larger, more functional multimers (including “ultralarge” multimers) are targeted to cytoplasmic storage granules for storage in the above-mentioned Weibel-Palade bodies and alpha granules. Secretion of the larger, more functional multimers is an active process that occurs by basal (unstimulated) and stimulated secretion pathways. The basal (unstimulated) secretion of VWF is largely responsible for circulating VWF. Stimulated secretion of VWF due to physiologic situations/agonists results in release of VWF into the vessel lumen via the apical surface of endothelial cells.
Agonists for VWF secretion include alpha-adrenergic agonists such as epinephrine, thrombin, fibrin, histamine, and the vasopressin analog DDAVP. (Note, DDAVP is used to treat mild VWD). This luminal secretion of large/ultra-large VWF multimers enhances VWF binding to platelets and may also increase the local concentration of VIII to enhance clot formation. Release of VWF from endothelial cells that are under shear stress (presumably turbulent blood flow due to endothelial injury) produces long strands of VWF that remain anchored to the endothelial cells by P-selectin. This allows binding of additional VWF molecules, platelets and ADAMTS13, the primary enzymes that breaks down VWF multimers. (As you recall from a prior discussion of TTP, aka thrombotic thrombocytopenic purpura, antibodies to ADAMTS13 cause a lower ADAMTS13 level, result in inability to break down VWF multimers, leading to clotting with thrombocytopenia.)
Clearance and control of VWF: The typical half-life of circulating VWF is about 8 to 12 hours. Interestingly, the rate of clearance of VWF is in part determined by glycosylation of the VWF protein, which is regulated by various transferases, including ABO blood group glycosyltransferases. Adults with type O blood have approximately 25 to 30% lower VWF levels than individuals with types A, B, or AB blood. This is due to a portion of the carbohydrate present on VWF that is similar to the blood groups and influences clearance of VWF.
Treatment of VWD:
- Desmopressin (DDAVP): This synthetic vasopressin that is used to treat patients with mild VWD (generally type 1 patients). It causes release of stored VWF to increase levels of circulating VWF. It doesn’t work in all types of disease, particularly in type 3 VWD and can be problematic in certain subtypes of type 2 VWD. The drug can cause hyponatremia, so free water restriction/fluid restriction is advised with significant use. It only works for a few doses, as stored levels that are depleted will require production of new VWF after they are released. Tachyphylaxis can be seen with treatment.
- VWF replacement: Plasma-derived products enriched for VWF (and factor VIII) or recombinant VWF can be given, and this is often used in more severe forms of VWD.
- Drugs that inhibit fibrinolysis can be used to stabilize clots/prevent clot breakdown and hence can help reduce bleeding.
- Hormonal therapy with contraceptives or a hormonal IUD can reduce bleeding from menses in females with VWD.
More Laboratory testing: Fibrinolysis
Alright, we have covered taking a bleeding history. We have mentioned that collagen vascular disorders/problems that result in bleeding have no good clotting lab tests and no treatments (this is basically a diagnosis of exclusion for a bleeding problem). We have covered in detail lab tests to assess for problems with secondary hemostasis (the clotting cascade), which generally include the PT, aPTT, and TT. We have covered in detail the lab tests to assess for problems in primary hemostasis involving platelets and VWF. We will now briefly review problems with fibrinolysis.
Fibrinolysis:
Fibrinolysis is the process by which clots are dissolved. If we do this too readily, we can run into bleeding problems. If we are deficient in fibrinolysis in some way and don’t dissolve clots, then we can have a clotting problem due to lack of clot lysis. However, bleeding problems are more common (than clotting) in disorders of fibrinolysis….
Review of clotting: The end of the clotting cascade: Fibrinogen is converted by Thrombin to Fibrin. The fibrin is then crosslinked by factor XIII to make the clot stronger. Then, when we are done making the clot, we eventually will want to dissolve it.
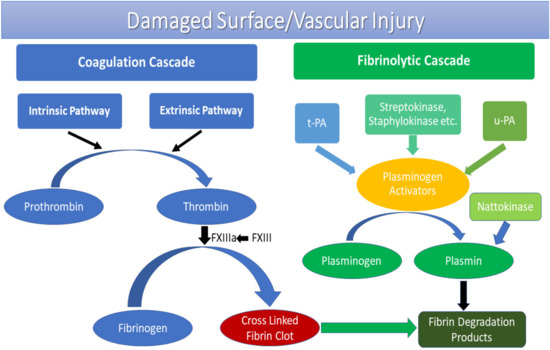
(Graphic courtesy of MDPI)
Fibrinolysis is the process where a fibrin clot is broken down or dissolved:
So, how do we break fibrin clots down? Fibrin is partially digested by Plasmin (recall that plasminogen is acted on by t-PA and u-PA to produce plasmin). This partially digested fibrin is then further broken down by plasmin into fibrin degradation products (FDP) and D-dimers. Fibrinolysis has natural inhibitors to slow clot lysis: Plasminogen Activator Inhibitor-1, alpha2-plasmin inhibitor (aka alpha2-antiplasmin), and Thrombin Activatable Fibrinolysis Inhibitor (TAFI).
Plasminogen activator inhibitor (PAI-1) inhibits t-PA, thus there is less conversion of plasminogen to plasmin by t-PA and clot is not broken down. If a person has a deficiency of PAI-1, t-PA is not inhibited leading to more plasmin production. This results in overactive clot lysis/fibrinolysis occurs and that person is prone to bleeding due to excessive clot lysis.
Alpha2-plasmin inhibitor (a2-PI) also inhibits plasmin degradation of fibrin. Hence, patients with a deficiency of a2-PI will have more active plasmin, leading to more active fibrinolysis and risk for bleeding.
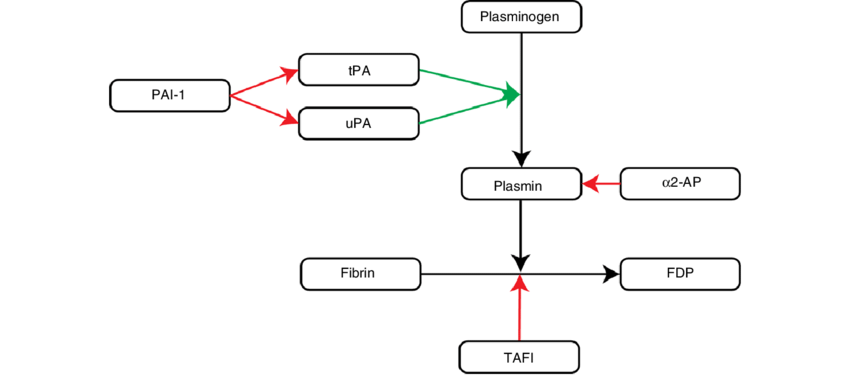
Disorders of Excessive Fibrinolysis that lead to bleeding:
Congenital: PAI-1 deficiency, a2-PI deficiency.
Acquired: Examples include end stage liver disease/cirrhosis, which results in reduced levels of a2-PI and impaired hepatic clearance of tPA.
Lab testing for fibrinolysis: You aren’t required to know this, so this is only if you are interested:
Typical lab findings in hyperfibrinolytic states include the following (note, we are talking about bleeding due to abnormal fibrinolysis only. One could argue that DIC involves hyperfibrinolysis, but DIC is a separate entity we have covered previously, and coag labs and platelets would be abnormal in DIC).
CBC/platelet count: Typically normal unless there has been significant bleeding to cause anemia.
Fibrinogen: Often low with significant fibrinolysis, because it is being consumed in the process of bleeding due to fibrinolysis with the body subsequently trying to form more fibrin to stop the bleeding.
PT and aPTT may be normal or prolonged. The mechanism of prolongation of the PT/PTT in hyperfibrinolytic states involves the degradation of fibrinogen and/or upstream coagulation factors and is way beyond the scope of what you need to know.
D-dimer: Increased due to fibin degradation.
Euglobulin clot lysis time: shortened. This is the classic test for overactive fibrinolysis, although it will also be shortened in patients with factor XIII deficiency—see next sentence.
Urea clot lysis testing—a test for fibrin cross-linking, which will be faulty in factor XIII deficiency: this should be normal with excessive fibrinolysis, unless the patient has a factor XIII deficiency.
Treatment of excessive fibrinolysis (this you do need to know):
Treatment involves drugs that are antifibrinolytics. These are lysine analogues that bind the kingle domain of plasminogen/plasmin, thereby leading to disruption of the interaction between plasmin and fibrin. There are 2 available agents in the USA, tranexamic acid and e-aminocaproic acid. These can both be given IV or PO.
Risks of using antifibrinolytics? Can you guess? Yes, clotting is the answer! These drugs reduce clot break down, so the risk of using these drugs is excessive clotting.
Authors and Contributors:
Corliss Newman, MD authored this course pack material based partly upon slide lecture material prepared by Jill Johnsen, MD. Nicholas Burwick, MD and Kristi Rice, MD provided editing assistance.