
8 Disseminated Intravascular Coagulation (DIC) and Thrombotic Microangiopathies (TMAs)
Student Learning Objectives
In a prior lecture, we covered disorders of primary and secondary hemostasis in the form of platelet disorders, Von Willebrand disease, and two types of hereditary hemophilias. There are some other disorders we’ll highlight here. These disorders generally present with low platelets, as well as with a distinct type of hemolysis (break down of RBCs) due to red cells being traumatically lysed in the vascular system, called microangiopathic hemolytic anemia. Disseminated Intravascular Coagulopathy (DIC) involves simultaneous bleeding and clotting problems. Thrombotic Microangiopathies are disorders that result in low platelets and clotting. These problems are BAD, so you don’t want to miss them in clinical practice! (Some folks think of DIC, disseminated intravascular coagulation, by another name: Death is Coming….)
Microangiopathic Hemolytic Anemia (MAHA)
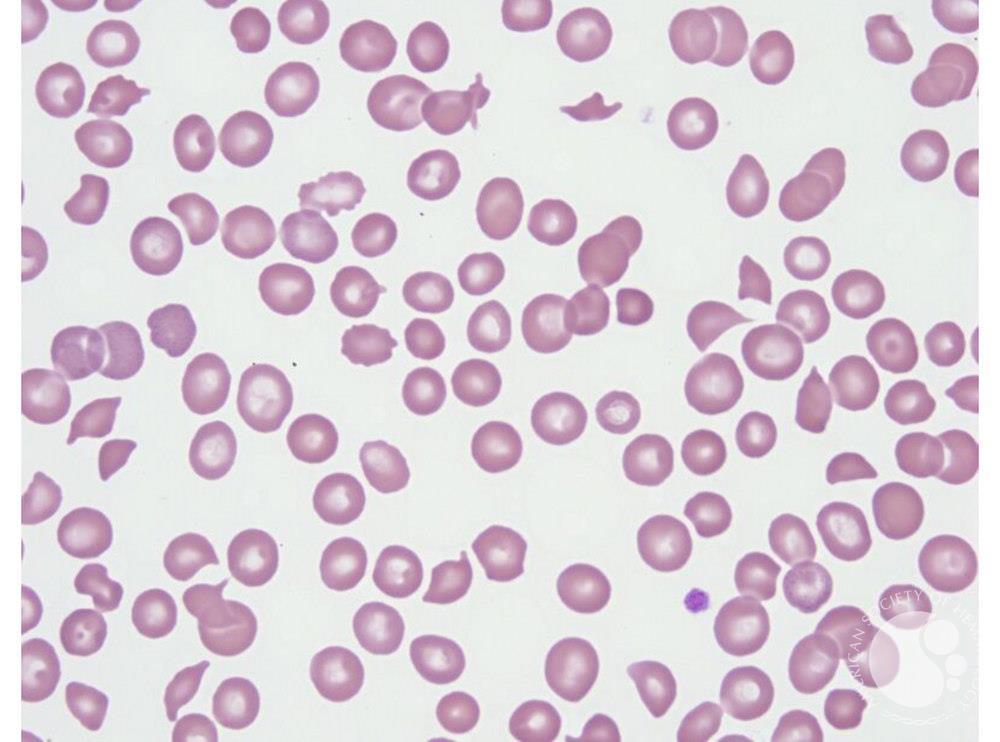
MAHA is a descriptive term for non-immune hemolysis (Coombs-negative hemolysis) that results in intravascular red blood cell fragmentation that produces schistocytes on the peripheral blood smear.
Causes of MAHA:
-
- Red cell trauma from prosthetic heart valve
- Red cell trauma from ventricular assist devices
- Disseminated Intravascular Coagulation (DIC)
- Thrombotic Microangiopathies (TMAs)
- Infections
- Other
Thrombotic Microangiopathies (TMAs) and DIC are types of MAHA that are associated with abnormalities in the microvasculature (small arterioles and capillaries) that also commonly cause activation and consumption of platelets.
Image Courtesy of ASH Image Bank
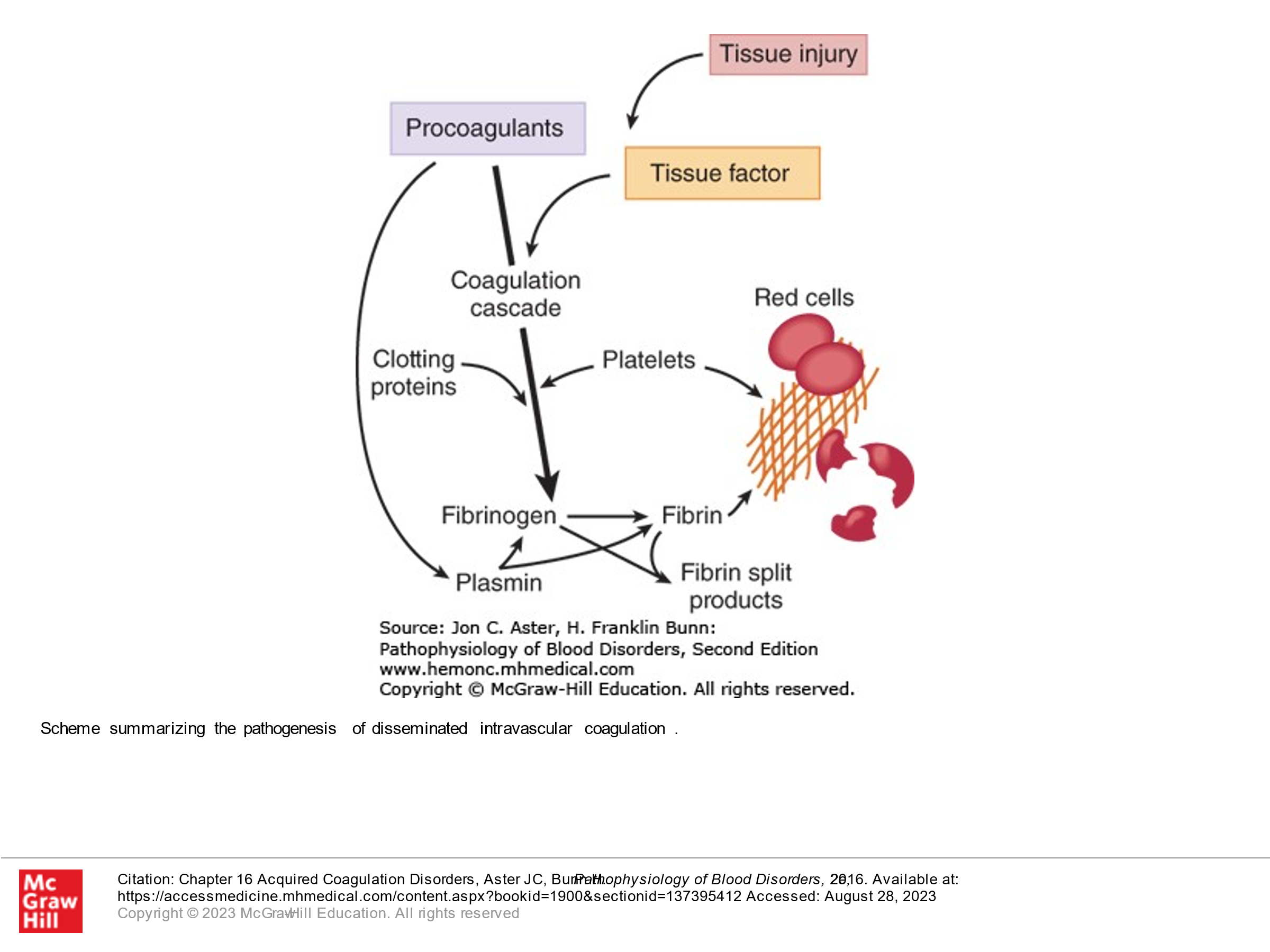
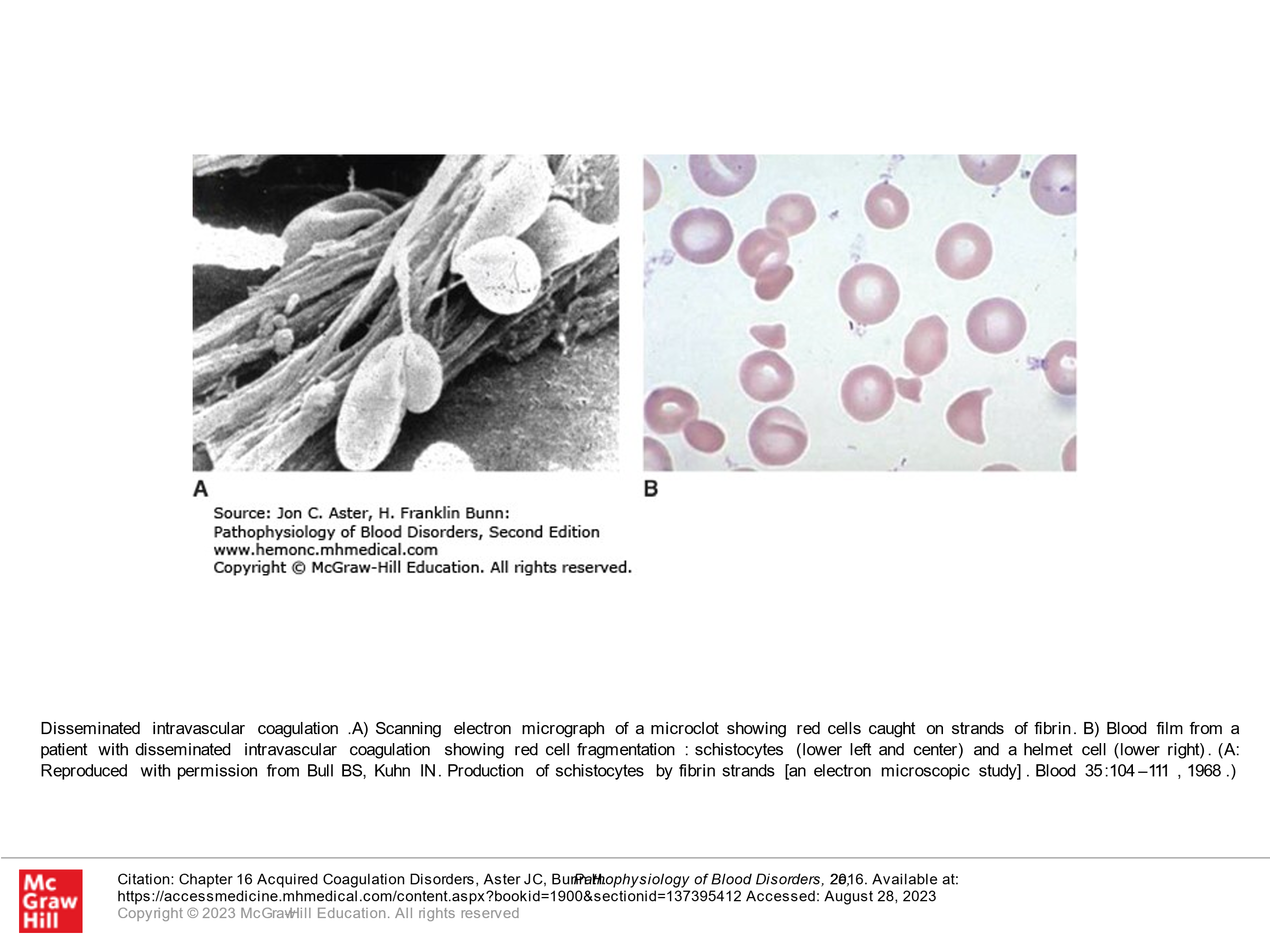
Disseminated Intravascular Coagulation (DIC):
Optional textbook reading: Part II Hemostasis and Thrombosis, Chapter 16 (Acquired Coagulation Disorders, section on DIC)
DIC, as its name describes, involves clotting occurring intravascularly throughout the body. This is a medical emergency and can result in bleeding or clotting (or both) in a patient and is life-threatening. It is thought that this occurs when blood is exposed to tissue factor (TF) or TF-like substances. This can occur in the setting of trauma, obstetric accidents/OB complications, diffuse vascular injury, increased endothelial permeability, activated monocytes, and via tumor cells—they may have their own protein that activates coagulation. DIC is a pathologic response to an injury which involves activation of blood clotting and fibrinolysis which are so robust that they are not controlled by the normal physiologic control mechanisms. With DIC we see abnormal clotting. Due to consumption of clotting factors and platelets activated by clotting, we can also see bleeding in patients with DIC, in addition to clotting. Laboratory findings in patients with DIC include the following:
- Elevated PT
- Elevated PTT
- Decreased platelet count
- Decrease in individual clotting factors
- Presence of fibrin degradation products ( D-dimers, FDPs).
- Presence of Schistocytes (fragmented RBCs) due to shearing of red cells on the intravascular clots.
Treatment of DIC generally involves treating the underlying cause and is otherwise supportive. If patients are bleeding, then factor support (platelets, plasma, etc.) is given. If patients are clotting, anticoagulation can be done, generally using heparin.

|
Vascular Damage Leading to Release of Tissue Factor |
|
Bacterial sepsis Gram-negative organisms Meningococcus Pneumococcus Gram-positive organisms Metabolic stress Acidosis Shock Heat stroke |
|
Release of Tissue Factor from Injured or Pathologic Tissue |
|
Obstetrical complications Placental abruption Retained placenta/fetus Placenta previa Amniotic fluid embolus Preeclampsia/eclampsia Malignancies Solid tumors, mucin-secreting adenocarcinomas Acute promyelocytic leukemia Severe burns |

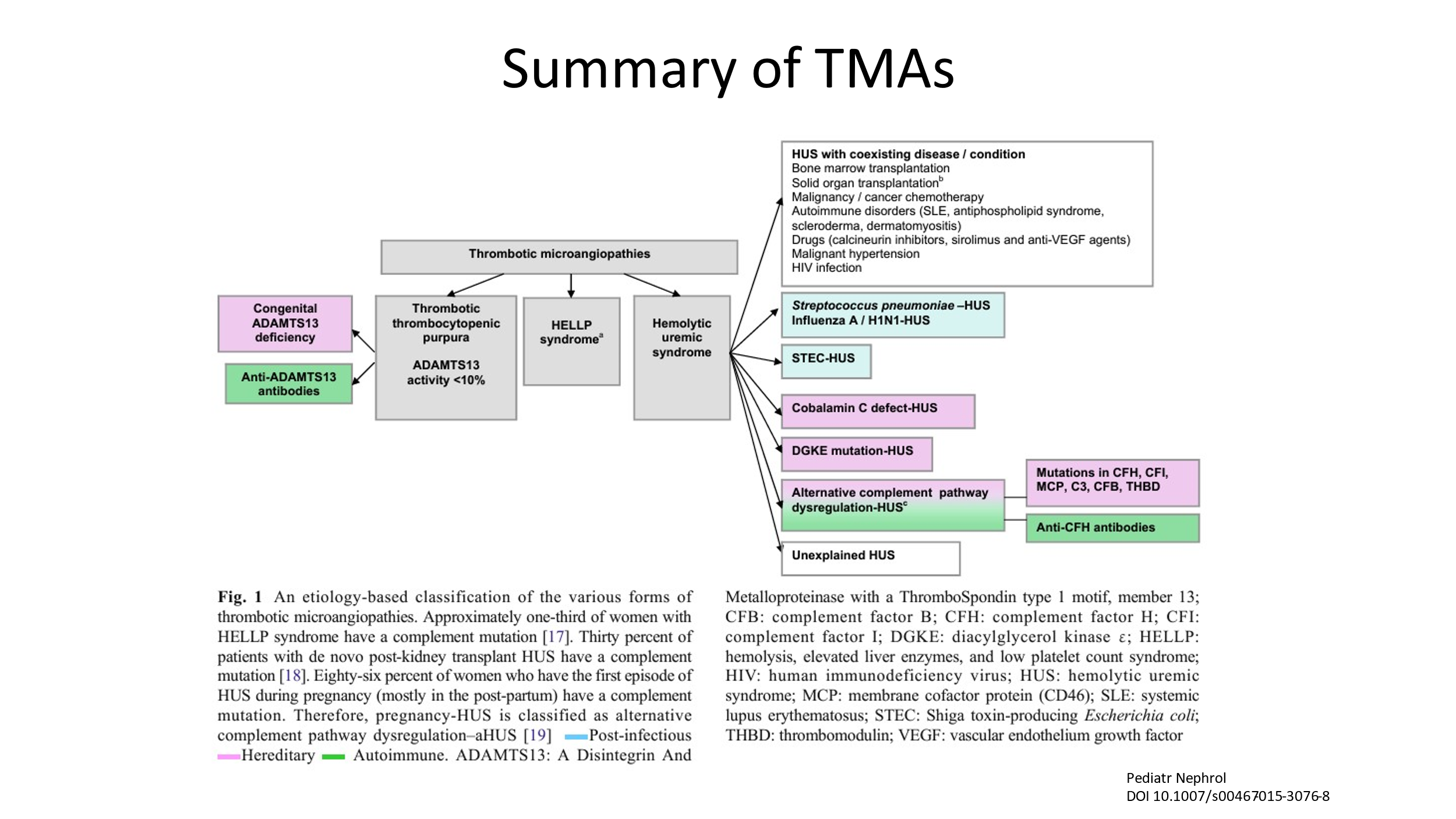
Thrombotic Microangiopathies (TMAs)
Thrombotic Microangiopathies are clinical syndromes defined by low platelets, fragmentation of red blood cells (schistocytes), and organ damage due to the formation of microscopic blood clots in capillaries and small arteries. Unlike DIC, clotting factors are not generally consumed and fibrinolysis is not significantly activated. Hence, unlikely DIC, the PT, PTT, and fibrinogen are normal (usually) in patients with TMAs.
There are several subtypes of TMA. Here are some as listed below.
- Thrombotic Thrombocytopenic Purpura (TTP)
- Underlying etiology of TTP is deficiency of ADAMTS13. This can be a hereditary deficiency of ADAMTS13 (hereditary TTP, upshaw-schulman) or an acquired deficiency of ADAMTS13 (acquired TTP, due to auto-antibodies against ADAMTS13).
- Shiga-toxin Hemolytic Uremic Syndrome (HUS) (previously was called “typical” HUS)
- Complement-mediated TMA (previously has been called “atypical” HUS)
- Coagulation-mediated TMA
- Metabolism-mediated TMA
- Drug-induced TMA
Note: Atypical hemolytic uremic syndrome (aHUS) is an older nomenclature that is no longer used. aHUS was historically used to describe children with MAHA, thrombocytopenia, and kidney failure not associated with diarrhea. The term aHUS is often used to describe any patient with a TMA who doesn’t have ADAMTS13 deficiency or a documented shiga-toxin. It is better to simply define the TMA as per the above list, as treatment will depend upon the classification.
You should have a good understanding and be able to distinguish between the following TMAs (as well as be able to distinguish DIC):
- TTP (Thrombotic Thrombocytopenic Purpura)
- Complement-mediated TMA
- Shiga-Toxin HUS
Thrombotic Thrombocytopenic Purpura (TTP)
- TTP is a thrombotic microangiopathy (TMA) caused by severely reduced activity of the von Willebrand factor-cleaving protease known as ADAMTS13 (activity <10%)
- TTP is characterized by small-vessel platelet-rich thrombi that cause thrombocytopenia, microangiopathic hemolytic anemia (MAHA), and sometimes cause end-organ damage.
- TTP is a medical emergency that is almost always fatal if appropriate treatment is not started promptly.
- With appropriate treatment, the survival rate is over 90%.
Recall from prior lectures that von Willebrand factor (VWF or vWF) is a glycoprotein that is critical to primary hemostasis in that it helps bind platelets to collagen when the vascular wall is damaged. (VWF is also important for the clotting cascade in that it binds and stabilizes factor VIII in circulation). VWF resides in plasma, in subendothelial matrix, and in storage granules in endothelial cells and platelets. VWF is a multimer composed of repeating subunits that create several binding sites for proteins/platelets that VWF interacts with. Production of VWF results in a variety of sizes of VWF multimers in circulating blood. As long multimers of VWF circulate, the shear stress of circulation will cause the VWF to unfold/elongate. This exposes the A2 domain of VWF, which is the cleavage site of ADAMTS13, an enzyme that regulates the size and pro-thrombotic activity of VWF. In normal circulation, as the VWF polymer/multimer elongates, ADAMTS13 (aka VWF-cleaving protease, aka vWF proteinase) binds to A2 and cleaves this area of the VWF to make shorter VWF multimers. This prevents unwanted binding of platelets to circulating VWF.
In TTP, there is a deficiency of ADAMTS13. Long VWF multimers are not cleaved as they elongate in circulation due to shear stress. This allows platelets to bind to these elongated VWF multimers and become activated.
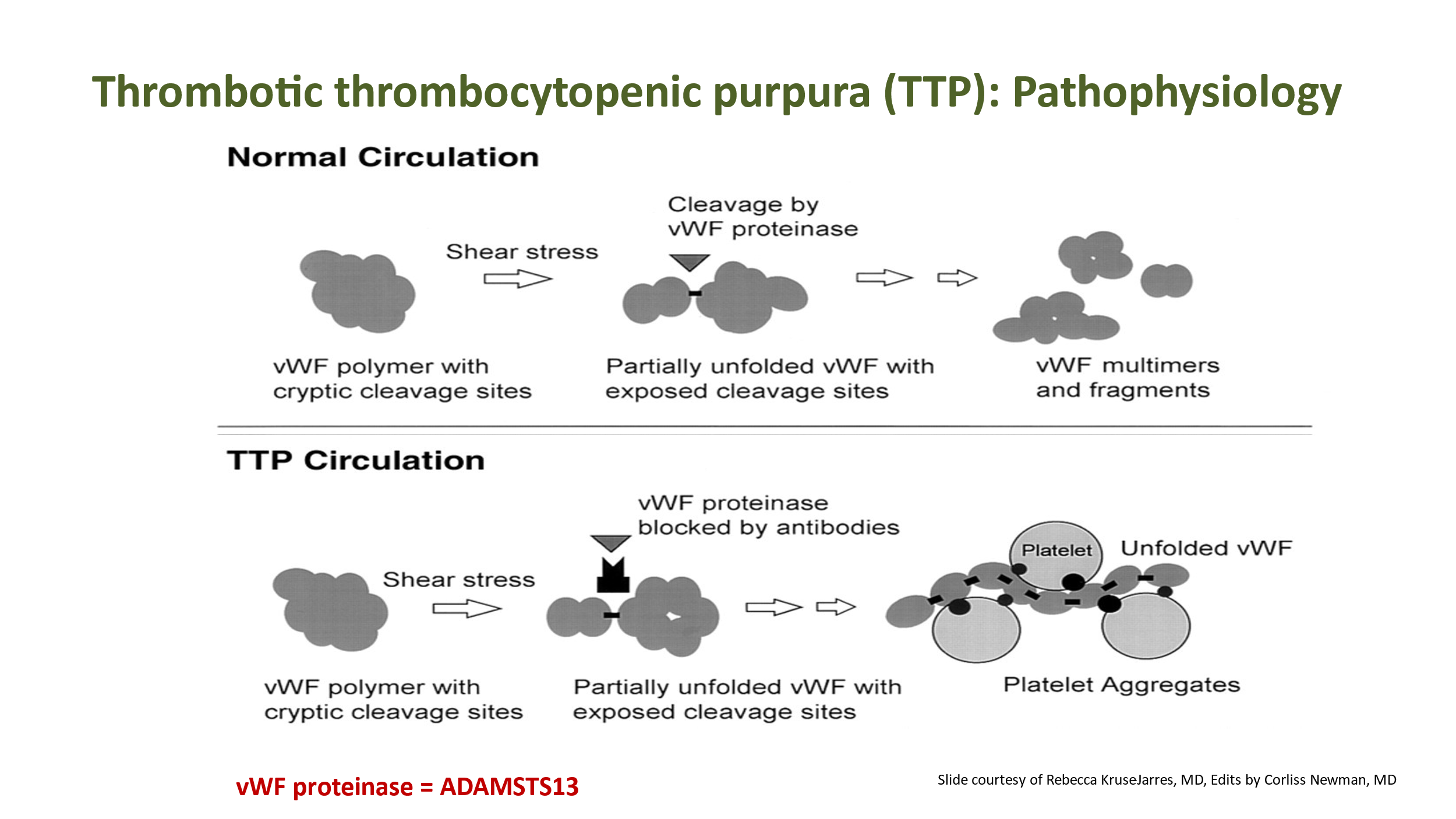
With platelets aggregating/activated with long VWF multimers, plugging in small vessels/capillaries occurs, resulting in end-organ damage. Typically, brain and GI tract are affected. However, any organ can be affected. Kidney function can be adversely impacted, although frank kidney failure is much less common with TTP than is seen with other TMAs. Fever is also common. Patients with TTP often present with fever, neurologic findings, GI symptoms, and some decreased renal function. Laboratory findings will show thrombocytopenia, microangiopathic hemolytic anemia (MAHA), normal PT/PTT, and often an elevated creatinine. The key diagnostic finding is decreased ADAMTS13 activity. Usually this is due to an inhibitor (often an auto-antibody against ADAMTS13), unless the patient has a congenital deficiency of ADAMTS13 (these patients are diagnosed young, in the setting of congenital deficiency). TTP is a medical emergency, and treatment must be started immediately, often before the ADAMTS13 level is known. Mortality rate is high if TTP treatment is not initiated quickly.
Treatment of TTP
Treatment of immune mediated TTP involves Therapeutic Plasma Exchange (TPE). TPE is the mainstay of TTP treatment. This is a procedure where the patient is placed on a pheresis machine and the patient’s own plasma is removed from their blood and exchanged with donor plasma.
TPE works in 3 ways:
- Large VWF multimers are removed
- Auto-antibodies to ADAMTS13 are removed
- Functional ADAMTS13 is provided in the normal donor plasma
In addition to therapeutic plasma exchange—other treatments such as glucocorticoids (eg prednisone), Rituximab (anti-CD20 monoclonal antibody) and Caplacizumab can also be considered. Prednisone is commonly given in conjunction with TPE. glucocorticoids and rituximab are immunosuppressive agents that reduce auto-antibody production. Caplacizumab, which is now being used more commonly, is a monolconal antibody that binds the A1 domain of Von Willebrand factor which inhibits the interaction of VWF with the glycoprotein Ib-IX-V receptor on platelets. Caplacizumab improves the platelet count and decreases TTP-related death, recurrence of TTP, and thromboembolic events.
In inherited TTP, TPE or immunosuppression is not required since there is not an auto-antibody. treatment is with plasma infusion to give back normal ADAMTS13in order to improve the deficient levels.
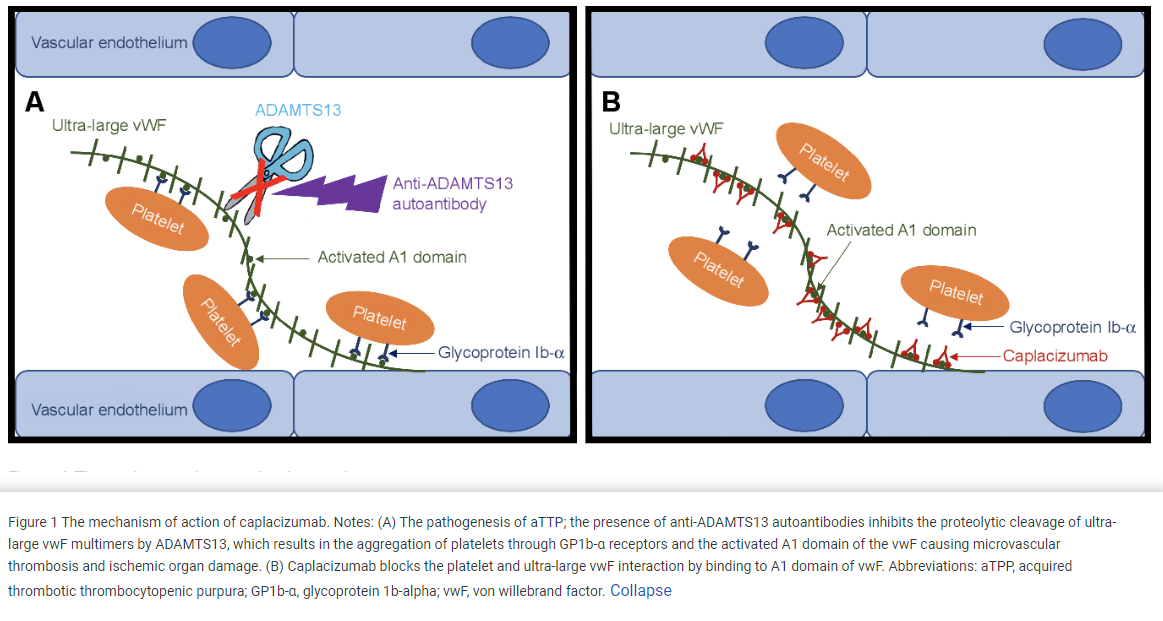
Published in Drug Design, Development and Therapy 2019
Caplacizumab as an emerging treatment option for acquired thrombotic thrombocytopenic purpura
Courtesy of Semantic Scholar
Hemolytic Uremic Syndrome
Hemolytic Uremic Syndrome got its name because this disorder typically had triad presentation with the following:
- Microangiopathic hemolytic anemia (MAHA)
- Thrombocytopenia
- Small vessel thrombi with a preponderance of damage seen to the kidneys.
There are a variety of types of HUS, and now these disorders are named by the inciting event/underlying problem. Note that TTP and HUS are both thrombotic microangiopathies (TMAs). TTP is very distinct from HUS, in that it TTP is due to loss of ADAMTS13 activity. Also, TTP tends to present with more neurologic and gastrointestinal symptoms, and generally doesn’t cause severe renal failure. the thrombocytopenia also tends to be more severe in TTP (<30), while in HUS it is often low but >30. HUS is characterized by severe kidney injury. The main 2 types/most common types of acquired TMAs that present with HUS are as follows:
Shiga-Toxin HUS (previously called typical HUS).
Complement-mediated TMA (aka complement-mediated HUS, previously called atypical HUS)
Shiga-Toxin Hemolytic Uremic Syndrome (formerly called “typical” HUS)
This is a disorder caused by Shiga-toxin producing E. coli HUS (0157:H7). Classically this is a disorder in children. It starts with a watery diarrhea that progresses to a bloody diarrhea, along with severe crampy abdominal pain and nausea and vomiting. In children who develop E. coli enterocolitis, approximately 15% will progress to development of ST-HUS. The pathogenesis of the this disorder appears to be when the Shiga-toxin binds to cell membrane glycolipid Gb3, is then internalized, and causes apoptosis of the affected cell. The Shiga-toxin has several additional effects on endothelial cells, one of which is enhanced expression of functional tissue factor—this may contribute to the microvascular thrombosis that is observed. Shiga-toxin causes damage or activation of endothelium, red cells, and platelets. Of note, cell membrane glycolipid Gb3 is strongly expressed on the glomerular endothelium, which might explain why kidney failure is the presenting problem with ST-HUS.
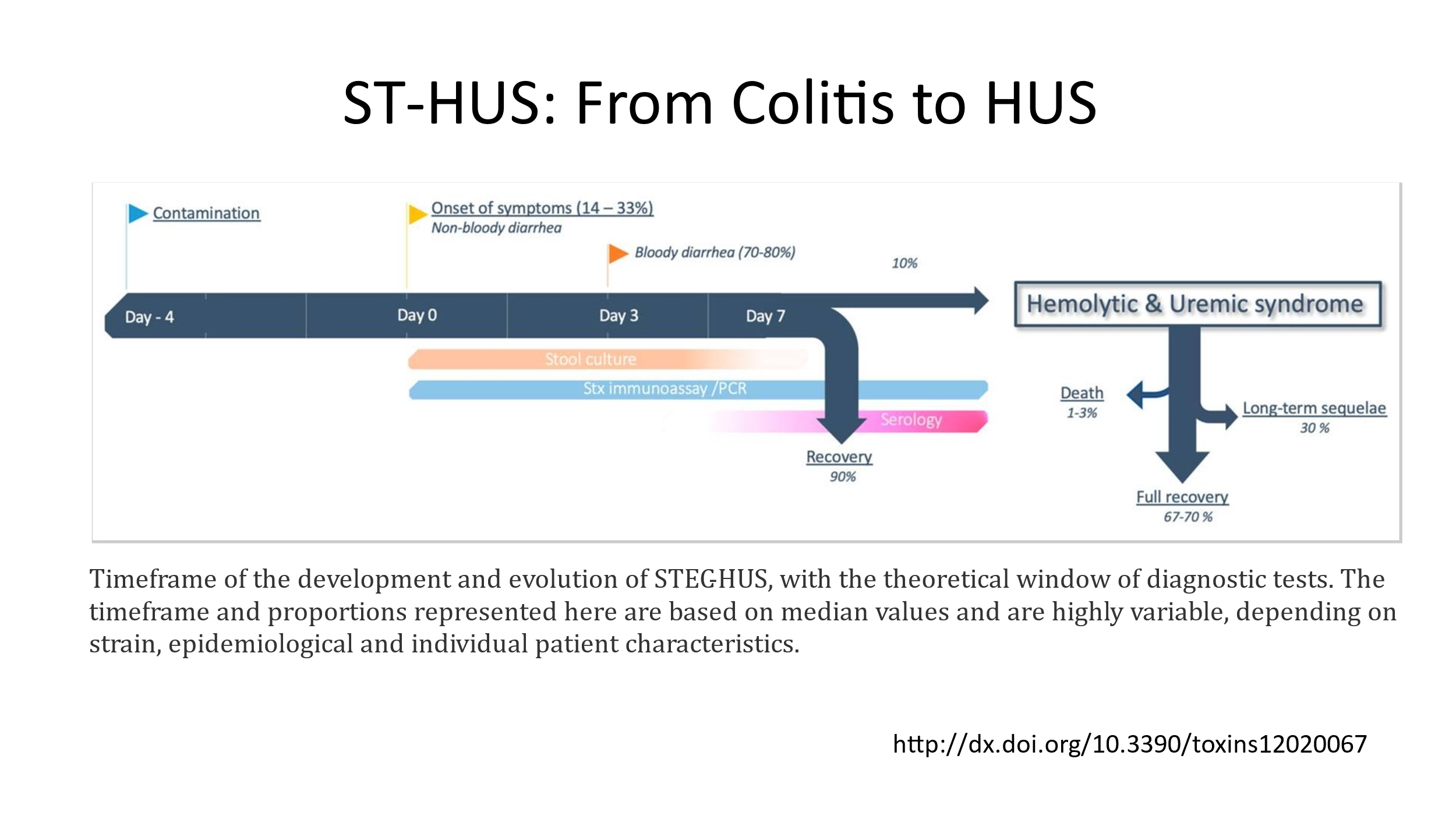
Treatment of ST-HUS is supportive care. Antibiotics are generally discouraged, as the infection is usually waning by the time the problem is diagnosed and there is some concern that antibiotic therapy could result in more release of Shiga-toxin. Anti-diarrheal medication is also discouraged, as they don’t help and may increase the risk for development of ST-HUS. NSAIDs should be avoided, as they can aggravate acute kidney injury. The mainstay of treatment is hydration and treatment of electrolyte disturbances. Transfusion of RBCs is often needed to treat anemia. Patients with HUS can become significantly anemic quickly. Platelets should not be given unless the patient has active bleeding. Dialysis may be needed if the patient has signs/symptoms of uremia, significant azotemia with BUN over 80, severe fluid overload, and/or severe electrolyte abnormalities that require dialysis to correct.
Complement-Mediated TMA (formerly called “atypical” HUS)
- Complement-mediated TMA (CM-TMA) is a TMA associated with inherited pathogenic variants in complement genes or acquired autoantibodies against complement factor H (CFH) or other such complement regulatory proteins.
- CM-TMA has associated triggers such as pregnancy, surgery, infections, malignant hypertension, and other inflammatory conditions; (these are all complement amplifying conditions)
- CM-TMA is thought to be a two-hit disorder: First hit is the inherited or acquired complement variants/acquired antibodies targeting complement by the patient. Second hit is the associated triggers that lead to complement activation: pregnancy, surgery, etc.
- 3 main mechanisms of complement dysregulation attributed to disease pathogenesis: loss of function mutations in complement regulatory proteins (e.g., Factor H, Factor I, Membrane Cofactor Protein/CD46, and thrombomodulin); gain of function mutations in effector proteins, such as Factor B or C3; and formation of neutralizing autoantibodies against Factor H.
- Family history of TMA, kidney failure, or unexplained kidney failure or death during pregnancy may be suggestive of an inherited CM-TMA.
- CM-TMA is driven by the generation of excess complement activation, often due to the loss of the natural regulators of the complement system such as complement factor H.
- Ultimately, dysregulated compliment activation leads to attacks on endothelial cells, particularly renal endothelial cells, causing damage leading to platelet activation and microangiopathic hemolytic anemia.
CM-TMA is treated differently than TTP or ST-HUS. The mainstay of treatment is anti-complement therapy, using C5 monoclonal antibodies such as eculizumab or ravulizumab. The diagnosis is made clinically in a patient who presents with symptoms more consistent with HUS and less likely TTP, doesn’t have a history of a diarrheal infection/is not a child, and has factors known to be associated with CM-TMA, such as pregnancy or recent surgery, etc. In this type of patient, testing of complement levels, testing for Complement Factor H antibodies, and Genetic testing of the 5 principal complement genes should be ordered. Immediately after these tests are drawn (before results are known), anti-complement therapy should be initiated. shiga toxin should be sent to rule out ST-HUS. Immunization against meningococcus should be given along with antibiotics against meningococcus due to the risk of infection with C5 complement blockade (impaired formation of the C5b-9 terminal MAC complex)
Summary: MAHA, DIC, TMAs
- MAHA (Microangiopathic Hemolytic Anemia) is a descriptive term for non-immune hemolysis (Coombs-negative hemolysis) that results in intravascular red blood cell fragmentation that produces schistocytes on the peripheral blood smear.
- MAHA and thrombocytopenia are seen in DIC (disseminated intravascular coagulation) and in TMAs (Thrombotic Microangiopathies).
- In addition to consumption of platelets due to platelet activation, the clotting cascade and fibrinolysis are also activated and unregulated in DIC, leading to consumption of clotting factors and fibrinolysis factors as well.
- TMAs do not have significant activation of the clotting cascade or fibrinolysis. Their hallmark is the presence of MAHA and thrombocytopenia in the absence of clotting abnormalities.
- TTP is a subtype of TMA due to depletion/loss of ADAMTS13 (VWF-cleaving protease), causing platelets to adhere to long VWF chains. Patients commonly present with neurologic symptoms, GI symptoms, and/or fever. Renal insufficiency, if present, is usually not severe. Therapeutic Plasma Exchange (aka plasmapheresis) is the mainstay of treatment. Caplacizumab, an anti-VWF antibody that blocks large VWF multimer binding, is approved to use with TPE in the treatment of TTP.
- ST-HUS is a subtype of TMA due to bacterial infection (usually E.coli 0157:H7) that produces shiga-toxin. This is a problem usually seen in young children, who often have bloody diarrhea when they are ill. The ST-HUS usually occurs after the initial infection has cleared. Renal failure/severe renal injury is common. Treatment is supportive.
- CM-TMA is a subtype of TMA due to dysregulated complement leading to endothelial cell damage, especially in the kidneys. Renal failure/severe renal injury is common. Treatment is to start anti-C5 monoclonal antibody therapy with eculizumab or ravulizumab.
Authorship: Course pack material written by Corliss Newman, MD. Information from slides created by Corliss Newman, MD and by Rebecca Kruse-Jarres, MD used in writing this course pack.