
8 Frontiers of genetic diagnosis and therapy
Session Learning Objectives and a quick synopsis:
Advances in genetics are rapidly and profoundly transforming medical diagnosis, treatment, and ethics. This session offers a brief overview of emerging genetic technologies, many of which have been recently introduced into practice.
SLO1Compare and contrast indications and limitations of different genetic tests
Cytogenetic tests are useful for evaluating for structural or copy number changes affecting a large number of genes when there are clinical indications that a patient may have a chromosomal abnormality. Evaluation of specific genes or use of large gene panels is appropriate when a differential diagnosis can be formulated. Previously undiagnosable, one-of-a-kind patients often benefit from whole exome or whole genome sequencing.
SLO2Compare and contrast sources of DNA used for genetic testing
DNA taken from cells representative of the patient’s germline constitution, easily obtained from blood or via skin biopsy, is useful for genetic testing. Tumor tissue can be used to identify cancer-specific mutations. DNA from tumor tissue can sometimes be detected in peripheral blood, urine, or feces. Amniotic fluid, placental tissue, or fetal DNA circulating in maternal serum can be used for prenatal genetic diagnosis.
SLO3Be prepared to explain possible outcomes of genetic test results to patients and their families
Genetic tests can return with a positive result, a negative result, a variant of undetermined/unknown significance, or a worrisome incidental finding unrelated to the problem that motivated the patient to seek clinical attention. Patients need to be informed of the potential outcomes, and physicians need to understand how to react to such findings.
Autosomal and sex-linked recessive disorders are amenable to “gene addition” forms of gene therapy. Autosomal dominant disorders require that the mutant allele be inactivated. In vivo and ex vivo therapeutic technologies involving viral delivery of genes, genome editing, and oligonucleotide modification of gene expression and mRNA splicing continue to evolve.
Main text
SLO1Compare and contrast indications and limitations of different genetic tests
Several technologies are in current use for performing genetic tests.
Sanger DNA sequencing. Sanger DNA sequencing (also known as dideoxy DNA sequencing, due to its use of chain-terminating dideoxynucleotide substrates) is sort of an analog technology in which many copies of identical molecular fragments of DNA are analyzed at the same time. There is a single readout of results, in the form of an electropherogram tracing. Sanger sequencing works best when there is a limited genetic differential diagnosis for the disorder being considered, such that only a few genes or a known mutation are being evaluated. For example, Sanger DNA sequencing can be used to confirm diagnosis of a suspected hemoglobinopathy, which is often first evaluated by hemoglobin electrophoresis.
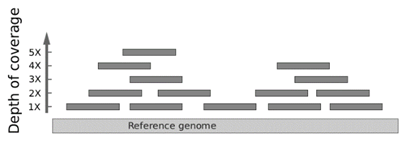
Next-generation DNA sequencing. Next-generation DNA sequencing is sort of a digital technology where many different DNA fragments can be more economically sequenced at once. The entire population of sequenced DNA molecules are evaluated individually, yet as an overlapping composite in order to interpret the sequence at any given base pair. Next-generation DNA sequencing can be used to evaluate multiple genes at once, including even the entire exome (i.e., the ~1% of the genome contained in exons) or whole genome. Because each nucleotide position is evaluated by multiple short DNA sequence reads to a high “depth of coverage” (or “read depth”), there is a potential to identify non-germline somatic mutations occurring in cancer or in mosaic disorders. For example, a single base position might be read more than 100 times. If the mutation is a heterozygous single base substitution, then, on average, about 50% of the reads should reveal the variant. If a variant base is found to occur at a significantly lower frequency, then that might be taken as evidence of mosaicism or clonal heterogeneity if studying a tumor.
Gene panels. Next-generation DNA sequencing is now invariably used for large gene panels. For example, retinitis pigmentosa, an inherited form of progressive blindness, is extremely genetically heterogeneous. There are more than 100 genes known to cause the disorder, so a gene panel test would be appropriate in this situation. To perform such a test, genomic DNA is hybridized to synthetic target sequences that capture relevant genes for analysis.
Exome sequencing. Next-generation DNA sequencing is the only technology available for analysis of the exome, which constitutes ~1% of the human genome or ~30 Mb (million base pairs), spread out across ~180,000 exons from ~20,000 total genes. Exome sequencing is appropriate when the disorder is so unique that there is literally no good guess as to what the patient may have.
Limitations of DNA sequencing. DNA sequencing has several limitations. Regardless of Sanger sequencing vs. next-generation DNA sequencing, if two different mutations are found in a particular gene known to be associated with an autosomal recessive disorder, it cannot be readily determined whether they are occurring in trans (one mutation on one parental chromosome and the other mutation on the other parental chromosomal homolog as would be required for autosomal recessive inheritance with each parent being a carrier) or in cis (both on the same parental chromosome with the other parental chromosome being normal). The only way to “phase” the distribution of variants discovered through DNA sequencing is to sequence the parents and determine how they segregate within the family or else resort to research studies involving the physical isolation of a single DNA molecule, which is generally outside the scope of readily available clinical laboratory testing. (There are still newer DNA sequencing technologies, capable of sequencing one molecule for long stretches at a time; however, currently cost and high error rates largely limit their use to the research setting. Nevertheless, they may be ready for the clinic soon.) For this reason, when performing exome sequencing on an individual where the mode of inheritance is unknown (and that is frequently the case since exome sequencing is usually reserved for patients where the diagnosis is uncertain), then it is best to sequence both parents (known as “trio” sequencing), if available, at the same time as the patient.
Neither Sanger sequencing nor next-generation sequencing is especially good at detecting insertion or deletion mutations, and next-generation sequencing is particularly poor at resolving short repetitive sequences such as those encountered with neurodegenerative disorders like the CAG repeat responsible for Huntington disease. Instead, for the latter, PCR with electrophoretic separation of the products is usually performed to simply evaluate the length of the repetitive tract.
Cytogenetic testing. In spite of the availability of next-generation DNA sequencing, there is a continuing need for cytogenetic tests such as karyotype, FISH, and microarray, which do not actually involve sequencing the genome directly. Only a karyotype can provide an overview of gross chromosomal abnormalities; however, its resolution is limited to changes to no smaller than a few million base pairs. FISH is not generally suitable for evaluating changes at a genome-wide level, unlike a karyotype or a microarray; in general, a clinical diagnosis must be entertained and specifically tested for. FISH is useful for confirming particular microdeletion or microduplication syndromes (i.e., copy number variant disorders). FISH is also useful for testing for particular tumor-specific abnormalities of diagnostic, therapeutic, or prognostic significance (such as the t(9;22) Philadelphia chromosome in CML). To a large degree, microarrays have supplanted FISH and offer a genome-wide perspective for evaluating for copy number variant disorders resulting from segmental duplications and deletions, such as 22q11.2 deletion syndrome. It is important to realize, however, that microarrays cannot detect so-called “copy neutral” changes such as balanced reciprocal chromosomal translocations or intrachromosomal inversions because, unlike a karyotype or even FISH, they match DNA sequences in the absence of visualizing the structure of the chromosome.
SLO2Compare and contrast sources of DNA used for genetic testing
Genetic testing requires DNA, and it can be obtained from a variety of sources.
Constitutional DNA. DNA representative of all the cells of the body and hence corresponding to the “germline” is conveniently obtained from peripheral blood (“peripheral” to distinguish it from blood cells obtained from the bone marrow), via venous phlebotomy, or from saliva. Sometimes dermal fibroblasts isolated by a skin biopsy are used in place of blood; one situation would be to screen for a germline mutation that might cause leukemia, where acquired mutations might also be present in white blood cells.
Fetal DNA. Fetal DNA can be obtained by amniocentesis (where fetal cells are directly sloughed off into the amniotic fluid) or by chorionic villus sampling (CVS), in which a placental biopsy is performed. More recently, screening tests employing next-generation DNA sequencing to analyze cell-free fetal DNA circulating in maternal serum have become feasible.
Cell-free DNA. As is the case during pregnancy where fragments of fetal DNA circulate in maternal serum, degraded DNA fragments are exuded by cells, including cancer cells, undergoing apoptosis or necrosis. There is active research into using next-generation DNA sequencing technology to sensitively detect cancer-associated mutations in cell-free DNA, obtained from serum, urine, or stool. Dubbed a “liquid biopsy,” the concept is that activating proto-oncogene or inactivating tumor suppressor gene mutations may be detectable in cell-free DNA at the earliest stages of cancer. An emerging challenge is to identify the tissue of origin of cell-free tumor DNA, based on tissue-specific epigenetic marks that may influence how the circulating fragments are partially digested.
SLO3Be prepared to explain possible outcomes of genetic test results to patients and their families
Variants of undetermined/unknown significance. How does the physician decide whether a variant is significant or causative? If the genetic change obviously disrupts protein-coding, then that it is a pretty good indication that the variant is deleterious. Any DNA sequence change that leads to a termination codon, such as a “nonsense” mutation involving substitution of a termination codon, or that alters exon/intron splicing stands a good chance of being deleterious. Assessing the significance of an amino acid missense substitution is more problematic. We can make educated guesses on the basis of whether the altered residue resides in a functional portion of the protein, whether the amino acid that is disrupted is evolutionarily conserved across species, and whether the amino acid substitution itself is nonconservative (e.g., acidic to basic sidechain). In fact, there are numerous formal algorithms (even meta-algorithms that combine analyses for several) for making these determinations, but they can only go so far.
Regardless of how a variant observed in a particular gene may disrupt a protein or its expression, it is also helpful to determine the frequency of the variant in the general population, as well as to review the literature or various databases to learn if that particular variant has been identified in patients previously. If it is rare in the general population but recurrently reported among patients, then it’s more likely to be causative of disease. It is also helpful to know if the variant segregates with disease in the family. For example, if a parent also has the same disorder and also possesses the variant, then that provides evidence in favor of its causality, but if the parent is unaffected then it more likely represents a benign polymorphism. If the variant is a de novo mutation (i.e., not found in either parent) and neither parent is affected, then that can be taken as very strong evidence in favor of causality. There just are not that many de novo mutations, so any new protein-coding change arising de novo is highly suspect.
Variants of undetermined/unknown significance (“VUS”), not surprisingly, can lead to much consternation on the part of the physician and the patient. It is particularly problematic for individuals whose ancestry is under-represented in control and disease databases, in which case distinguishing between a pathogenic mutation vs. a benign polymorphism becomes even more challenging.
Incidental findings. Genetic testing is increasingly moving toward large gene panels and exome sequencing because many disorders can be caused by a large number of genes. It may be more economical to simply perform an exome analysis rather than to devise and continually revise an ever-growing list of genes responsible for a particular clinical phenotype. Yet, by casting a wider net, the number of identified genetic variants grows larger.
Consider a case where exome sequencing is performed on a child with a clinically undiagnosed disorder of intellectual disability and congenital anomalies. Exome testing returns with a positive result and leads to a diagnosis. However, unexpectedly, exome sequencing identifies a pathogenic mutation in a gene predisposing to cancer, such as a heterozygous mutation in TP53, responsible for Li-Fraumeni syndrome. Should these results be reported? What if the parents expressly said at the time testing was performed that they did not desire to learn about anything else lurking in the genetic data? One could conceivably argue that since there are no clear data that early cancer detection in Li-Fraumeni syndrome improves clinical outcome, there is little harm in not revealing this information. But what if, instead, there was an incidental finding of a heterozygous mutation in a gene, such as KCNQ1, encoding a voltage-gated potassium channel that causes long QT syndrome, an inherited disorder predisposing to lethal cardiac arrhythmias? Sudden cardiac death can be avoided by implantation of an automatic defibrillator.
Controversy surrounds the issue of return of incidental genetic findings to patients. One criterion for distinguishing whether a particular incidental finding should or should not be reported is whether it’s “actionable” (i.e., something could be done about it, whether that be screening for cancer or implanting an automatic defibrillator). Currently, one professional body, the American College of Medical Genetics, identifies 49 genes, KCNQ1 and TP53 included, which if incidentally found to contain a mutation on genetic testing (in particular, exome sequencing) performed for other indications should be returned to the patient (or the parents of a child), regardless of stated desires.
It is therefore important that patients be informed of the possibility that genetic testing could lead to identification of variants of undetermined significance or incidental findings that no one may have been expecting.
Gene therapy. Gene therapy refers to the genetic manipulation of the somatic genome for the treatment of diseases.
It is important to emphasize that targets chosen for gene therapy are somatic cells, not those contributing to gamete formation. In fact, at least for now, a deliberate effort is made to avoid modification to sperm or eggs, in order to prevent permanent changes to the human genome, thereby forever altering the future of our species.
Recently, troubling news came from China, where doctors employed CRISPR-based genome editing (a method described further below) to purposefully introduce the CCR5-Δ32 mutation into human embryos toward the goal of conferring HIV resistance and increasing intelligence (the latter being another reported association for this variant). Children alive today are said to have resulted from these experiments. Those responsible received worldwide opprobrium. Some of myriad concerns include the possibility that CRISPR will introduce mutations in other genes (“off-target” effects) and that there may be untoward consequences of introducing even genetic variation thought to have favorable effects. The moral implications of manipulating the human germline are profound.
While much of the focus of gene therapy has been directed at the treatment of inherited disorders, ongoing efforts address acquired disorders, such as cancer or HIV infection. Gene therapy is a field very much in flux, as new gene delivery systems and genome-editing technologies continue to evolve. Recent years have seen success, but the field is fraught with hyped claims, a rush to proceed while sidestepping safety concerns, financial conflicts of interest, and bad behavior inside and outside of the laboratory and clinic. For our purposes, treatment of a topic of such wide scope is necessarily brief. We will review some of the general principles guiding the selection of diseases amenable to gene therapy and methods for gene delivery and genome modification.
Treatment of inherited disorders. A first consideration is what genetic disorders might be appropriate for gene therapy. Autosomal recessive and sex-linked recessive disorders, in general, make for suitable targets for “gene addition” approaches to gene therapy because they usually result from a reduced or absent amount of activity of the gene product. Many metabolic disorders are simply due to an absence of the enzymatic activity required for catalyzing a step in a metabolic pathway. Therefore, supplying a normal copy of the deficient gene could conceivably make up for insufficient activity resulting from reduced amounts of protein encoded by the mutant gene. In contrast, for an autosomal dominant disorder, the mutation is heterozygous, meaning that the cell already possesses a wild type copy of the gene. Consequently, simply introducing a normal copy of the gene, in order to generate greater levels of wild type protein, will not work in the same way that it will for a recessive disorder. Instead, approaches to gene therapy for an autosomal dominant disorder depend on inactivating expression of the mutant allele.
The next consideration is whether to target cells in vivo, as opposed to isolating cells from a patient, modifying them in vitro, and then returning them to the patient—a so-called “ex vivo” approach to gene therapy. It would be ideal if therapeutic gene delivery could occur entirely in vivo. Unfortunately, to achieve therapeutic correction in a target tissue, a fairly large number of cells need to be modified. A tractable target for gene therapy is to perform ex vivo modification of hematopoietic stem cells in which a patient’s own cells, once therapeutically altered, can be autologously re-engrafted into the patient’s bone marrow. Of course, this is primarily of benefit for disorders involving the bone marrow.
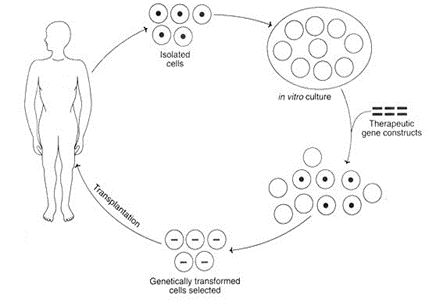
The following sections on gene therapy introduce many specific examples of a disease and approaches being developed for treatment. The important take-away is to recognize how understanding of genetics offers new opportunities for designing therapeutic measures. The actual diseases and proposed details for therapy are less important than the concepts and logic that you can anticipate seeing in future treatments.
Gene therapy viral vectors. Most vectors used to introduce a foreign gene, or to modify an endogenous gene, are based on viruses.
Early on, retroviruses were introduced for gene therapy. A disadvantage to retroviral vectors is that there is a risk that the retrovirus will disrupt a gene at the site of insertion into the host genome and thereby lead either to that gene’s loss of activity or to inappropriate expression. If it were a tumor suppressor gene or proto-oncogene then one could imagine that this may lead to cancer. In fact, this risk has materialized in several high-profile trials of gene therapy involving treatment of two different sex-linked recessive immunodeficiency disorders, X-linked severe combined immunodeficiency (X-SCID), in which there are few T and NK (natural killer) lymphocytes, and Wiskott-Aldrich syndrome, characterized by eczema, thrombocytopenia, immune deficiency, and bloody diarrhea. In those trials, enhancer elements of the retrovirus activated expression of proto-oncogenes at the site of their integration, curing the immunodeficiency but causing leukemia.
Adeno-associated virus (AAV) is a single-stranded DNA virus that requires adenovirus as a helper virus to productively infect cells. The native AAV has an unusual property in that it tends to integrate into the targeted human cell’s genome specifically at a certain locus on chromosome 19. However, AAV modified for use as a vector has lost this site specificity. AAV vectors infect non-dividing cells. At present, AAV has attracted much attention for both ex vivo and particularly in vivo gene therapy because it appears to provide prolonged and moderately high levels of expression of the therapeutic gene. The size limit of the gene that can be packaged is somewhat smaller than that which can be delivered by retroviruses and adenovirus. Its single-stranded genome may also make it valuable in genome editing strategies that make use of targeted homologous recombination.
Happily, after decades of toil, the dream of gene therapy has finally come to fruition and has moved beyond the research setting, making gene therapy-based cures of genetic disorders broadly available to patients. The first FDA (United States Food and Drug Administration) approved gene therapy products employ AAV vectors. Multiple clinical trials targeting other diseases and utilizing other vector technologies are rapidly moving forward.
Leber’s congenital amaurosis is a genetically heterogeneous congenital form of blindness. One genetic etiology results from homozygous mutations in the gene encoding RPE65, a retinal enzyme contributing to the “photo cycle” by regenerating visual pigment involved in the detection of light by rods and cones. FDA-approved gene therapy consists of subretinal injection of an AAV vector expressing RPE65 cDNA—a “cDNA” is a compact, intronless version of the gene retrotranscribed from its mRNA.
Spinal muscular atrophy is an autosomal recessive degenerative neuromuscular disorder most commonly caused by homozygous mutation of SMN1, a gene encoding a transcription factor-associated protein required for motor neuron survival. It can now be treated with intravenous infusion of an AAV vector expressing SMN1. Its current cost is $2 million. Shortly following its recent FDA approval, allegations of data manipulation surfaced, and top drug company executives were fired.
Oligonucleotide-based therapies targeting RNA. An oligonucleotide is a short chemically-synthesized polynucleotide. While their use would not cure disease in the sense that they permanently edit the genome or otherwise add back a normal copy of the mutant gene, they are capable of treating a disease by modulating the expression of a mutant gene. In the following strategies, RNA complementary to gene transcripts forms short stretches of double-stranded RNA that block translation and promote degradation of the transcript or that modulate RNA splicing:
RNA interference (RNAi). RNAi encompasses both naturally occurring complementary microRNA (miRNA) and artificial (such as short hairpin RNA (shRNA)) gene regulatory systems, which utilize the cell’s RNA-induced silencing complex (RISC) as well as other nucleases to, somewhat durably, repress gene expression. This strategy can be used to silence the expression of deleterious, typically dominantly acting, mutations or to inactivate viral gene expression. Several RNAi-based drugs have been FDA-approved. The first such drug is used to treat transthyretin amyloidosis, which results from mutations in the transthyretin retinol transporter that misfold and form amyloid deposits in heart, brain, and other tissue. Two other RNAi-based oligonucleotide drugs are now in use to treat hypercholesterolemia. One silences expression of apolipoprotein B, a component of low-density lipoprotein (LDL) cholesterol produced in the liver and thereby directly lowers LDL levels by preventing its formation. The other targets the mRNA encoding proprotein convertase subtilisin-kexin type 9 (PCSK9), an enzyme negatively regulating levels of the LDL receptor. The insight for why reducing levels of PCSK9 should be therapeutic came about from genetic studies showing that people with PCSK9 mutations that reduce its activity have lower LDL levels.
Exon-skipping therapies. Somewhat similar are therapies employing chemically modified DNA-based oligonucleotides that can re-direct the cellular splicing machinery to skip exons containing deleterious mutations. For example, in Duchenne muscular dystrophy, some mutations in the gene encoding dystrophin create frameshifts or nonsense codons. Dystrophin is a gigantic gene, spanning over two million base pairs, containing 79 exons, and taking RNA polymerase 16 hours to transcribe. If the exon containing the mutation is deleted from the final transcript, by splicing around it during maturation of the mRNA, even though there may be a large in-frame deletion, the overall protein is still sufficient to function mostly normally, compared to the effect of the mutation which may be to either truncate the protein at the point the mutation occurs or, even worse, cause the entire transcript to decay due to nonsense-mediated decay. A therapy utilizing this approach to specifically target frameshift mutations in exon 51, by skipping over it so that exon 50 splices to join exon 52, is now FDA-approved. This therapy will not, however, work for patients with mutations elsewhere in the gene.
 Exon inclusion therapies. The gene, SMN1, whose mutations are responsible for spinal muscular atrophy as noted above, has a nearly identical and adjacent gene paralog, SMN2, resulting from an evolutionary gene duplication event. SMN2 differs by just a single translationally silent nucleotide substitution that alters its splicing. As a result, SMN2 is alternately spliced, skipping an internal exon required for full activity, and ordinarily only produces small amounts of intact functional protein, identical to SMN1. The number of SMN2 copies in the genome varies between zero and eight among different people in the population, due to highly polymorphic copy number variation in this region. It was observed that disease severity for patients with SMN1 mutations is inversely proportional to the number of copies of SMN2, meaning that residual expression of full length SMN2 can compensate for SMN1 mutations. Based on this observation, an FDA-approved therapeutic strategy was designed in which an oligonucleotide targeting an intron blocks the splicing signal that causes the exclusion of the exon in SMN2, thereby therapeutically increasing expression of full length SMN2, producing a protein identical to SMN1.
Exon inclusion therapies. The gene, SMN1, whose mutations are responsible for spinal muscular atrophy as noted above, has a nearly identical and adjacent gene paralog, SMN2, resulting from an evolutionary gene duplication event. SMN2 differs by just a single translationally silent nucleotide substitution that alters its splicing. As a result, SMN2 is alternately spliced, skipping an internal exon required for full activity, and ordinarily only produces small amounts of intact functional protein, identical to SMN1. The number of SMN2 copies in the genome varies between zero and eight among different people in the population, due to highly polymorphic copy number variation in this region. It was observed that disease severity for patients with SMN1 mutations is inversely proportional to the number of copies of SMN2, meaning that residual expression of full length SMN2 can compensate for SMN1 mutations. Based on this observation, an FDA-approved therapeutic strategy was designed in which an oligonucleotide targeting an intron blocks the splicing signal that causes the exclusion of the exon in SMN2, thereby therapeutically increasing expression of full length SMN2, producing a protein identical to SMN1.
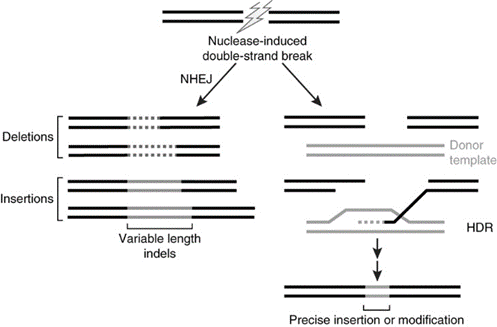
Genome editing. The human genome can be site-specifically modified. The general approach is to employ an engineered endonuclease (a DNase that cleaves internal to the linear molecule). The broken DNA molecule can then repair itself, usually by nonhomologous end-joining (NHEJ). When the broken ends are ligated back to together, a small deletion or insertion of variable length usually is generated. That by itself may be sufficient to, say, inactivate a dominant allele for an inherited disorder.
But, the approach can be taken even further. If another short piece of DNA is provided during the repair process, it can be employed as a substrate for directing the repair of the broken strand, through a process known as homology-directed repair (HDR). By supplying the appropriate template for homology-directed repair, humans can write what they want into the genome.
The breakthrough achieved with CRISPR is that DNA binding specificity is directed by an RNA template that guides the endonuclease to the cut-site where genetic modification is desired. Oligonucleotides can be rapidly, efficiently, and cheaply synthesized. The protein that binds the guide RNA and also cuts the DNA is a modified form of a naturally occurring bacterial protein, Cas9, that normally functions as an RNA-guided endonuclease. It seems that nature had evolved this technology in bacteria first, as a sort of molecular immune system against bacteriophages and DNA-based molecular parasites. Even newer technologies employ modified forms of CRISPR to edit bases directly in DNA or RNA. Targeting the mitochondrial genome has been experimentally demonstrated. Some of the editing machinery can be delivered as mRNA, using the same technologies as involved with COVID-19 vaccination.