
6 Cancer genetics
Session Learning Objectives and a quick synopsis:
Most cancer is not inherited. Instead, it usually results from bad luck caused by the accumulation of mutations acquired with aging or environmental exposures such as tobacco. These mutations happen throughout the genome and are generally inconsequential except when they disrupt certain important genes influencing cell growth. There is also a role for complex inheritance of risk factors for most malignancies. Nevertheless, some families do have inherited forms of cancer. The genes responsible for inherited forms of cancer are sometimes also mutated in non-inherited (sporadic) forms of cancer.
SLO 1Describe how a proto-oncogene transforms into an oncogene
Most proto-oncogenes are ordinarily involved in regulating cell growth. Mutations that unleash a proto-oncogene’s cancer-inducing potential increase its activity, such as through its over-expression. Only one allele needs to be mutated. Proto-oncogenes primarily undergo somatic mutations. Only in rare circumstances can they be tolerated in the germline (and consequently all cells of the body) and cause a hereditary cancer predisposition syndrome.
SLO 2Explain the two-hit model of tumor suppressor gene inactivation
In contrast to proto-oncogenes, tumor suppressor genes tend to exhibit a broader range of activities influencing cell growth. Tumor suppressor genes require loss of their activity in order to promote cancer. Consequently, most mutations inactivate them, and both alleles must be mutated to promote cancer. In inherited cancer predisposition syndromes, one allele is inactivated through a germline mutation, and inactivating mutation of the second allele is the rate-limiting step that causes cancer (the “two-hit” mechanism). Some of the same genes responsible for inherited cancer predisposition syndromes are also biallelically mutated in sporadic cases of cancer, occurring in individuals without an inherited germline cancer predisposition; however, in this setting both alleles are mutated somatically.
SLO 3Explain how inherited DNA repair deficiency leads to cancer
Inherited or acquired deficiency of DNA repair factors and other genes required to maintain the integrity of the genome is a cause of cancer. Nevertheless, mutations in DNA repair factors, by themselves, do not promote cell growth disturbances characteristic of cancer. However, a deficiency of DNA repair factors can lead to an accumulation of mutations throughout the genome, including in proto-oncogenes or tumor suppressor genes that do directly regulate cell growth, thereby leading to cancer. Many of these genes also fit into the category of “tumor suppressor” genes, requiring two mutations; when inherited, one allele is mutated in the germline and the other is acquired and restricted to the tumor, whereas, when sporadic, both alleles are mutated somatically.
Main text
Cancer involves mutation of genes.
There are two generally accepted categories of genes whose mutation gives rise to cancer: oncogenes (known as “proto-oncogenes” before they are mutated) and tumor suppressor genes. Genes involved in DNA repair or that otherwise contribute to the integrity of the genome and gene expression are also frequently mutated in cancer; many of these also behave as tumor suppressor genes, at least with respect to the two-hit mechanism.
The conceptual framework for describing cancer genes and much of the terminology in use today is intricately tied to the experiments through which they were discovered.
SLO 1Describe how a proto-oncogene transforms into an oncogene
Discovery of proto-oncogenes. Oncogenes were originally recognized as agents that produced cancer in animals and that could be isolated from viruses with an RNA genome. These viruses—and other “retroviruses” like them—contain an RNA genome and possess the enzyme, “reverse transcriptase,” that reverse transcribes their RNA genome into DNA upon infection. Retroviral oncogenes represent a mutant version of a gene contained in the host genome and that the virus has usurped. The non-mutated form of the gene, that is found in the host organism, is known as a “proto-oncogene.” Later it was appreciated that somatic mutation of proto-oncogenes can lead to 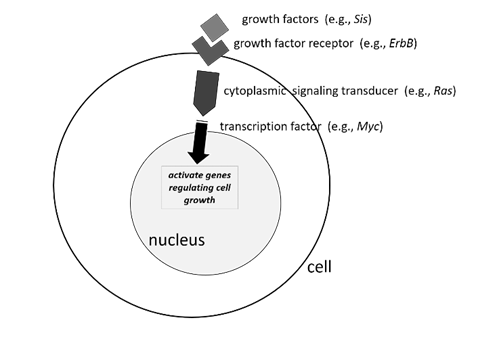 human cancers that are not associated with retroviral infections.
human cancers that are not associated with retroviral infections.
Many mammalian proto-oncogenes are known. In contrast to tumor suppressor genes (as we will soon discuss), the mutations that transform a proto-oncogene into a cancer-causing oncogene act dominantly at the cellular level. That is, one mutant copy (allele) of the oncogene is sufficient to perturb cell growth, and the wild type allele of the proto-oncogene is insufficient to halt the process.
Proto-oncogenes can be tied to cellular pathways regulating growth and differentiation. Mutations that transform a proto-oncogene into an oncogene render the protein it encodes hyperactive. Typically, this is a signaling protein involved in relaying an extracellular signal such as a growth factor to gene regulation in the nucleus.
For example, RAS, frequently mutated in many human tumors, encodes a GTPase that is “switched on” by incoming receptor signals and undergoes GTP/GDP exchange. Activated RAS protein ultimately turns on downstream genes involved in cell growth, differentiation, and survival. (Humans have three closely related RAS genes, HRAS, KRAS, and NRAS, which we will collectively and nonspecifically refer to here as just, “RAS.”) Cancer-associated, activating RAS mutations, which transform the proto-oncogene into an oncogene, cause the protein to be constitutively switched on all the time, even in the absence of receptor signals. Another frequently mutated human proto-oncogene is MYC, encoding a transcription factor that is situated downstream of multiple cell signaling pathways and activates a transcriptional program governing cell growth and division. In humans, oncogenic MYC mutations may lead to its overexpression or prevent its normal proteolytic turnover. Mutations in genes regulating any number of steps in cell signaling—levels of a particular growth factor, activation of its receptor, or conveyance of that signal downstream through the cytoplasm and into the nucleus—can lead to unregulated cell growth, a hallmark of cancer.
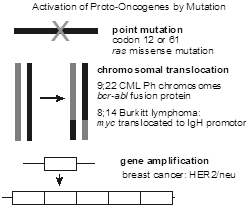 Several different types of mutations can transform a proto-oncogene into an oncogene. These include point mutation, chromosomal translocations that lead to gene fusion events, chromosomal translocations that lead to over expression by placing the proto-oncogene under the inappropriate control of another gene’s promoter or enhancer sequences, and gene amplification in which a gene is tandemly duplicated several times, thereby increasing its expression.
Several different types of mutations can transform a proto-oncogene into an oncogene. These include point mutation, chromosomal translocations that lead to gene fusion events, chromosomal translocations that lead to over expression by placing the proto-oncogene under the inappropriate control of another gene’s promoter or enhancer sequences, and gene amplification in which a gene is tandemly duplicated several times, thereby increasing its expression.
Somatic chromosome 9;22 reciprocal translocation, the so-called “Philadelphia chromosome,” is characteristic for chronic myeloid leukemia (CML). It results in a fusion of the protein-coding sequences of the BCR and ABL proto-oncogenes. It generates a fusion protein with a new tyrosine kinase activity. A highly effective tyrosine kinase inhibitor imatinib (trade name, Gleevec) was developed to target the BCR-ABL CML-specific tyrosine kinase.
Several different chromosomal translocations juxtapose various proto-oncogenes with the immunoglobulin heavy chain promoter on chromosome 14 and define certain subtypes of non-Hodgkin lymphoma (e.g., t(8;14) in Burkitt lymphoma).
Somatic amplification of the HER2 gene, encoding the EGF receptor 2 (EGFR2), in breast cancer is a therapeutic target of the monoclonal antibody-based drug, trastuzumab (trade name, Herceptin, among others).
Unlike tumor suppressor genes, mutations in proto-oncogenes are only rarely inherited. One such inherited cancer predisposition syndrome resulting from germline (inherited) transmission of a mutant proto-oncogene is multiple endocrine neoplasia 2 (MEN2), in which inherited mutations of the RET proto-oncogene can cause a syndrome with a high frequency of medullary thyroid carcinoma, parathyroid adenoma, and/or pheochromocytoma of the adrenal and related tissue. RET also frequently undergoes activating somatic mutation in individuals who sporadically develop medullary thyroid cancer in the absence of an inherited syndrome such as MEN2.
Characteristics of oncogenes
- Identified as transforming genes of animal retroviruses.
- An activated form of a cellular gene (proto-oncogene).
- Act dominantly at the cellular level, which means only one allele need be mutated.
- Mutations are somatic and seldom inherited (one exception is RET in MEN2).
SLO 2Explain the two-hit model of tumor suppressor gene inactivation
Tumor suppressor genes are genes whose normal function reduces the chances of cancer. Most tumor suppressor genes have been identified from mutations in individuals exhibiting autosomal dominantly inherited predisposition to cancer.
Knudson “two-hit” model of tumorigenesis. The prototype tumor suppressor gene is the RB gene, first isolated as the cause of familial retinoblastoma. In 1971, Alfred Knudson proposed the elegant “two hit” model of tumorigenesis to explain the epidemiology of retinoblastoma. Retinoblastoma is a childhood tumor of the retina. Knudson observed that about 40% of patients with retinoblastoma had an onset in infancy or early childhood. Those cases also tended to be bilateral, more frequently had a parent who also had retinoblastoma, and were often associated with additional malignancy later in life. In contrast, the remaining 60% of cases had a later age of onset, tended to be unilateral, almost never had a family history of the tumor, and were not associated with risk for additional types of tumors. Knudson correctly hypothesized that the 40% of cases with early onset, bilaterality, and risk for other tumors represented a germline mutation in a particular gene, whereas the 60% of cases with later onset, unilaterality, and no risk of secondary tumors resulted from somatic mutations of the same gene. He coined the term “tumor suppressor gene” for this hypothesized gene.
Knudson’s reasoning went something like this: Let’s define a tumor suppressor gene as one that, must be inactivated in order to cause cancer—unlike an oncogene (which had actually not yet been discovered) which must be activated by mutation to cause cancer. Since there are two copies of all autosomal genes, then we should require that both copies of a tumor suppressor gene must be inactivated in order to promote tumor development. Let’s say the chance of an inactivating mutation in any particular gene (here the tumor suppressor gene) in any particular cell is one in a million (10‑6). What is the probability of mutating both alleles of the same gene in any particular cell? It’s just 10-6 10-6, which equals 10-12. That’s a very small number, and it’s not likely that this will happen. In fact, even if there were a million cells, say in the retina, the probability that one of these cells sustains two mutations in the alleles of same gene—2 hits—is only one in a million. But, what happens if someone inherits a mutation that inactivates one copy of that gene, such that all of the cells of the body, including in the retina and other tissues, have, from the moment of conception onward, already sustained the first hit? Then the probability of the second hit is still one in a million per cell. But, since there are about a million cells in the retina, the chance that the second hit will occur is pretty large—so large, in fact, that retinoblastomas often form during embryonic development and are present at birth. That is what happens with autosomal dominant inheritance of an inactivating 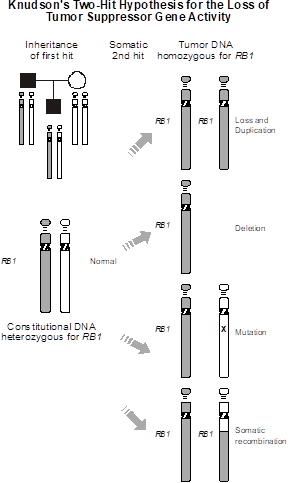 mutation in a single copy of the RB gene, whereas somatic mutation of both alleles of RB is a rarer occurrence. We predicted the chances of the latter happening to be about one in a million, and that is about how often retinoblastoma actually is found in the general population (i.e., among people who do not possess a germline mutation in one copy of RB).
mutation in a single copy of the RB gene, whereas somatic mutation of both alleles of RB is a rarer occurrence. We predicted the chances of the latter happening to be about one in a million, and that is about how often retinoblastoma actually is found in the general population (i.e., among people who do not possess a germline mutation in one copy of RB).
The rest of the differences between sporadic and inherited forms of RB is well explained: The occurrence of multiple tumors in individuals inheriting constitutional mutations in RB is because a second hit occurs with high probability. In fact, the second hit is a random event. Some unfortunate patients will therefore develop two or three or more retinoblastomas, while a few lucky individuals will be non-penetrant and never develop a tumor. The elevated risk for other types of tumors in individuals inheriting constitutional mutations in RB results from the fact that all of the cells in the body harbor the first hit mutation. Since loss of RB function can lead to the development of tumors in other tissues, individuals with inherited mutations in RB are also at high risk for other types of cancer. Finally, since tumor development requires two somatic mutation events in individuals not inheriting a mutant RB gene, it makes sense that the longer one lives, the greater the probability of exposure to mutation; this explains the later age of onset of tumor development in non-inherited forms of retinoblastoma.
Since the time of Knudson’s work, the RB gene has been identified. RB regulates transit through the cell cycle, by complexing with transcription factors. Somatic mutations of RB occur in many different types of tumors, and it turns out to be one of the most important genes in regulating orderly growth and differentiation of tissues.
Some individuals without a family history of retinoblastoma can still have a constitutional mutation in one allele of RB. These individuals represent new mutations—not surprisingly, given the poor genetic fitness of a childhood cancer predisposition syndrome. Such a person is subject to all the risks that befall an individual with a hereditary form of retinoblastoma, including the probability of 1/2 of transmitting it to each of their children.
Note that, in contrast to oncogenes, RB and other tumor suppressor genes act recessively at the cellular level. Only when function is completely lost, through mutations that inactivate both alleles, is tumor growth promoted.
Remember that proto-oncogenes must gain an activity to become a full-fledged oncogene. Tumor suppressor genes, in contrast, must lose their activity to cause cancer.
Most of the mutations of tumor suppressor genes (either inherited or somatic) are deletions or point mutations that cripple their function. Deletions of tumor suppressor genes are probably most common. Consequently, there is a unique experimental signature of a mutagenic event in the genome that takes out a tumor suppressor gene. The name for this is “loss of heterozygosity (LOH),” based on the fact that inactivating mutations of tumor suppressor genes frequently involve deletions or disomy for the mutant allele, which reveal themselves by LOH for polymorphisms that can be used to experimentally distinguish the two alleles.
Following are but a few of several examples of clinically important cancer predisposition syndromes resulting from autosomal dominant inheritance of heterozygous germline inactivating mutations in a particular tumor suppressor gene.
TP53 and Li-Fraumeni syndrome. TP53 is a DNA binding protein whose expression is induced by DNA damage. It was formerly, and often still, referred to as “p53” based on an apparent mass of 53 kilodaltons. Among other things, it regulates decisions regarding programmed cell death (apoptosis). One molecular mechanism to reduce the probability of developing a tumor is to kill off cells that have sustained extensive mutation. TP53 integrates numerous signals monitoring the health of the cell. When faced with overwhelming DNA damage, TP53 will make the decision to commit programmed cell death, in order to avoid malignant transformation of the cell. But, what happens if TP53 isn’t working? One can imagine that cells destined to form tumors will then not be eliminated.
Heterozygous germline mutations of TP53 are the cause of Li-Fraumeni syndrome, named for the two physicians who first characterized the disorder. Li-Fraumeni syndrome is an autosomal dominant disorder involving the inheritance of multiple malignancies, especially breast cancer, sarcomas, brain tumors, leukemia, and adrenocortical carcinoma.
Like RB, TP53 is somatically mutated in many malignancies. In fact, it is thought to be the single most commonly mutated gene in all of cancer.
Some families that would appear to have Li-Fraumeni syndrome but lack germline mutations in TP53 have been found to harbor heritable mutations in other genes similarly regulating the cell cycle and DNA repair, including CHEK2, encoding a protein kinase forming a DNA damage and replication “checkpoint.”
Neurofibromatosis 1. Neurofibromatosis 1 (NF1, also known as Von Recklinghausen disease) is a common autosomal dominant inherited syndrome of benign tumors known as neurofibromas occurring in conjunction with other benign tumors of the peripheral and central nervous system. The tumors can occasionally undergo malignant transformation, and the spectrum of malignancy includes neurofibrosarcoma, schwannoma, glioma, pheochromocytoma, and leukemia. Most of the time, though, the tumors remain benign and cause problems related to CNS “mass effects” and painful, disfiguring, or functional impingement of peripheral nerves. There are obvious cutaneous stigmata of the disease: the characteristic fleshy neurofibromas, hyperpigmented “cafe-au-lait” macules on the skin, raised hamartomatous lesions of the iris (Lisch nodules), and axillary and inguinal freckling (places where even freckled people don’t usually have very many). NF1 is caused by inactivating heterozygous germline mutations in the synonymous NF1 gene, encoding neurofibromin, a GTPase-activating protein (GAP) that regulates RAS activity. Loss of NF1 activity leads to constitutive activation of the RAS signaling pathway. Mutations in a number of other genes encoding components of the RAS activation pathway can produce inherited disorders with clinical overlap to NF1 and are collectively referred to as “Rasopathies.”
von Hippel-Lindau Syndrome. von Hippel-Lindau syndrome (VHL) is an autosomal dominant disorder of benign vascular tumors of the cerebellum, elsewhere in the brain and retina, pheochromocytoma, and renal cysts transforming to renal cell cancer. Even though they are benign, these vascular tumors cause problems when they enlarge and/or bleed in the brain and retina. As is the rule with most inherited tumor suppressor gene syndromes, both alleles of the gene (also called VHL) responsible for familial VHL are nearly invariably mutated in sporadic, non-familial forms of renal cell carcinoma.
Interestingly, mutation is not the only molecular mechanism for inactivating a tumor suppressor gene. It turns out that in some individuals with renal cell carcinoma, a CpG-rich region in the VHL gene promoter undergoes cytosine hypermethylation, switching off transcription of VHL and thereby leading to loss of expression, even without a frank mutation occurring in the DNA sequence. Somatic promoter hypermethylation (an epigenetic phenomenon, distinct from mutation) is a relatively common mechanism for inactivating a variety of tumor suppressor genes in sporadically occurring tumors.
The methylation status of a gene is fairly stable through cell division cycles. That is, once a gene is hypermethylated and switched off (whether it should occur aberrantly during tumorigenesis or in a developmentally appropriate manner during tissue differentiation, imprinting, or X chromosome inactivation), the methylation state is generally preserved in the two daughter cells resulting from mitotic cell division. A somatic change in the methylation status of the cell, however, is not preserved during meiosis; thus, the name “epigenetic,” as these states are, with possible rare exception, not heritable from one generation to the next.
The VHL protein is involved in signaling the tissue response to hypoxia.
Familial adenomatous polyposis. Familial adenomatous polyposis (FAP) accounts for about 1% of colon cancer. Probably all colon cancers begin as a benign polyp, which subsequently acquires additional mutations activating proto-oncogenes and inactivating tumor suppressor genes as it progressively transforms into a full-blown malignancy. Individuals with FAP usually have their colons carpeted with polyps by no later than their teenage or early adult years. Adenocarcinoma of the colon is therefore inevitable, and prophylactic colectomy in the early adult years is the only treatment option, as there are simply too many polyps to surveil and resect, even with frequent colonoscopy. The gene causing FAP is known as APC (for adenomatous polyposis coli), a protein with a complex role in cell cycle progression and extracellular communication and matrix attachment. Loss of APC activity results in chromosomal instability (CIN), leading to accumulation of multiple gross cytogenetic abnormalities of chromosome structure in tumor cells, which in turns leads to widespread activation of proto-oncogenes and inactivation of tumor suppressor genes.
As is the established paradigm with tumor suppressor genes, APC is commonly mutated in sporadic non-inherited forms of colon cancer.
Hereditary breast and ovarian cancer syndrome. Breast cancer is a common disease, and about one in nine of all females in North America will develop it. Breast cancer is inherited with complex genetics. As with other malignancies, most breast cancers appear sporadically, due to common genetic variants with weak effects, environmental exposures, and accumulation of somatic mutations as we age. A minority of cases, however, is familial and results from highly penetrant single-gene mutations.
Hereditary breast and ovarian cancer syndrome accounts for about 5% of all breast cancer. Familial breast cancer is genetically heterogeneous. BRCA1 and BRCA2 were the first two responsible genes to be identified.
In general, familial forms of cancer exhibit several hallmarks, which help differentiate it from coincidental family clustering of sporadic cases, including earlier age of onset and multiple primary tumors.
Thus, for hereditary breast and ovarian cancer syndrome, there tends to be a younger age of onset of breast cancer (often premenopausal, whereas most sporadic breast cancer occurs post-menopause). Bilateral occurrence of a tumor or multifocal occurrence of more than one primary tumor on a single side (just as with inherited forms of retinoblastoma) or multiple tumors arising over time (“metachronous”) are all more common in hereditary breast and ovarian cancer syndrome, as well as in other familial cancer predisposition syndromes. The presence of ovarian cancer in a family, which is much rarer than breast cancer, is often suggestive of families inheriting BRCA1 and, to lesser extents, BRCA2 mutations. The occurrence of male breast cancer, a rare event, is associated with BRCA2 and, to a lesser extent, BRCA1 mutation. Even individuals from breast cancer families who turn out not to have inherited the predisposing gene can still get breast cancer as a sporadic disease, just because breast cancer is so common in the general population.
Females inheriting a mutation in BRCA1 or BRCA2 face a lifetime risk of breast cancer of about 50-80%. Males inheriting BRCA2 mutations are at elevated risk for developing breast cancer (approximately 6% lifetime risk). Females inheriting a BRCA1 mutation have a lifetime risk of ovarian cancer of approximately 25-50%, whereas the risk is somewhat lower with BRCA2 mutations. Germline mutations in both BRCA1 and BRCA2 confer smaller elevations in risk for other types of tumors, including prostate cancer in males, melanoma, and pancreatic cancer.
Some populations show founder effects for BRCA mutations. About 2% of people of Ashkenazi Jewish European ancestry are heterozygous for one of three different mutant alleles of either BRCA1 (two different alleles) or BRCA2 (one allele). So large is this effect that most familial breast and ovarian cancer in this population can be attributed to one of these three mutations. Other ethnically isolated populations show a similar preponderance of founder mutations. For example, hereditary breast cancer frequently results from the same BRCA2 mutation in Iceland, where meticulous genealogical records trace it back to a Viking.
Genetic testing for familial BRCA1 and BRCA2 mutations is revolutionizing the approach to this disease. Females found to inherit a germline mutation in either gene can be screened more conscientiously and consider risk reducing therapies, including prophylactic mastectomy and/or oophorectomy.
BRCA1 and BRCA2 encode components of a large protein complex that detects and repairs DNA damage. The complex is referred to as the “Fanconi complex” because many of its components were first discovered as a cause of the genetically heterogeneous autosomal recessive disorder of DNA repair deficiency, known as “Fanconi anemia”.
Heterozygous germline loss of function mutation of BRCA1 and BRCA2 cause autosomal dominantly inherited adult onset of cancer (breast, ovarian, and, to lesser extents, other types of cancer), whereas homozygosity for the very same loss of function mutations in BRCA1, BRCA2, or other genes encoding components of the Fanconi DNA repair complex cause autosomal recessively inherited childhood onset of cancer (Fanconi anemia). Can you think of a reason why there is a difference in the ages of onset depending upon whether the mutations in these genes are heterozygous or homozygous in the germline?
Here’s why: Heterozygous mutations of BRCA1 or BRCA2 lead to half normal levels (haploinsufficiency) of BRCA1 or BRCA2 proteins. For most proteins, half-normal levels are good enough. BRCA1 and BRCA2 don’t directly regulate cell growth, division, or other cancer cell properties. And half-normal levels of BRCA1 or BRCA2 do not markedly impair DNA repair activities of the Fanconi complex. However, once a second hit (i.e., acquired somatic mutation) occurs in the remaining wild type allele of BRCA1 or BRCA2, then DNA repair is severely disrupted because there is no normal BRCA1 or BRCA2 protein left, at all. Consequently, the cell that now has absence of BRCA1 or BRCA2 loses DNA repair proficiency and accumulates somatic mutations at a much higher frequency throughout the genome. Mutations activating proto-oncogenes or inactivating other tumor suppressor genes arise and therefore ultimately produce a tumor. It should be easy to imagine that if someone is conceived with complete deficiency of BRCA1 or BRCA2 (as a result of autosomal recessive inheritance of homozygous mutations), that total loss of this DNA repair pathway occurs immediately and persists throughout life. Moreover, unlike the case for a second hit, which occurs randomly and only affects an occasional cell, every cell in the body will have lost this type of DNA repair capability. Consequently, mutations begin to accumulate in all cells throughout the genome, including in proto-oncogenes and tumor-suppressor genes directly responsible for disrupting normal growth characteristics in a cancer cell. It should be no surprise, then, that cancer can begin in childhood.
Heterozygous germline BRCA1 and BRCA2 mutations are responsible for most families with hereditary breast and ovarian cancer syndrome. Nevertheless, many inherited cancer gene syndromes are now known, and it is challenging to clinically distinguish among them. As a result, clinical testing for hereditary breast and ovarian cancer and other types of inherited cancer syndromes has moved away from testing single genes. Instead, clinical genetic testing is moving to large gene panels that apply next-generation DNA sequencing technologies to simultaneously evaluate for germline mutations in dozens of genes associated with hereditary cancer predisposition syndromes, including breast cancer.
Characteristics of tumor suppressor genes
- Identified as genes responsible for autosomal dominant human tumor syndromes.
- Recessive at cellular level, means both alleles must be inactivated.
- Often, the same tumor suppressor gene inherited in familial forms of the tumor is somatically mutated in non-hereditary, sporadic forms of the same tumor.
SLO 3Explain how inherited DNA repair deficiency leads to cancer
Mutations of proto-oncogenes and tumor suppressor genes lead to cancer, but what would happen if, rather than starting with a mutation in one of those genes, you began with a mutation in a gene responsible for maintaining the integrity of the genome? As we have discussed, we might expect that mutation of DNA repair and cell cycle control genes could lead to a cascade of mutations, eventually activating proto-oncogenes and inactivating tumor suppressor genes.
There are basically two types of genes in this category. One set of genes is directly involved in sensing and repairing DNA damage. The DNA molecule frequently undergoes physical damage: Bases are lost from the phosphodiester backbone, bases become chemically modified by exposure to carcinogens and reactive oxygen species, a DNA strand may break, and DNA polymerase can mistakenly insert the wrong base, leading to a “mismatch.” Much of this damage can be directly repaired, and that job falls to this first category of genes. The second major category of gene is involved in recognizing that DNA damage has occurred in the cell, temporarily halting the cell division cycle until the damage can be repaired, and then making an assessment as to whether the cell was fixed properly and should either re-enter the cell cycle or commit to programmed cell death. 2001 University of Washington Nobel Laureate Leland Hartwell in his seminal studies of yeast cell division mutants that failed to recognize when DNA damage had occurred introduced this latter “checkpoint” concept. A checkpoint is a temporary arrest in the cell cycle at which a particular list of safeguards is monitored before going any further.
(Recently, “checkpoint inhibitor” drugs targeting immune checkpoints have come into use for pharmacologic immunotherapy of cancer. These are completely different from cell cycle checkpoints, and the similar terminology should not be confused.)
Several genes involved in DNA repair and regulation of the mitotic cell cycle have been implicated as having a role in initiating sporadic and hereditary cancer.
Lynch syndrome. Lynch syndrome involves susceptibility to numerous types of malignancy, most commonly of the colon and elsewhere in the gastrointestinal tract, the endometrium of the uterus, and the ovary. It is a genetically heterogeneous disease inherited in an autosomal dominant fashion as a result of heterozygous inactivating mutations in one of several genes. The two most common genes responsible for Lynch syndrome are MSH2 and MLH1, with each being mutated in about a third of families with Lynch syndrome. Less often, Lynch syndrome is caused by mutations in MSH6 and PMS2. Proteins encoded by MLH1, MSH2, MSH6, and PMS2 are all part of a multi-protein complex involved in DNA double-strand mismatch detection and repair.
DNA mismatches represent incorrectly synthesized base pairs that result from misinsertion of the wrong base by DNA polymerase. Genomic instability of repeated sequences, known as “microsatellite instability,” is a hallmark of tumors from patients in Lynch syndrome families. Lynch syndrome accounts for about 5-10% of all cases of colon cancer overall.
Lynch syndrome, as well as hereditary breast and ovarian cancer syndrome, really blur the boundaries between tumor suppressor gene disorders and DNA repair deficiency disorders. Both syndromes are inherited in an autosomal dominant disorder. Both are due to heterozygous germline loss of function mutation in a tumor suppressor gene. Both require complete loss of activity through a somatic second hit through a two-hit hypothesis (with an inherited germline mutation and acquired, somatic mutation restricted to the tumor). Loss of DNA repair activity then leads to accumulation of secondary mutations activating proto-oncogenes and biallelically inactivating tumor suppressor genes.
Autosomal recessive syndromes of deficiency of DNA repair. The first group of DNA repair genes found to be involved in hereditary cancer predisposition syndromes is composed of rare autosomal recessive syndromes of malignancy. They are each characterized by a cellular deficiency in various aspects of DNA repair. The following lists some representative examples of diseases from this category.
Fanconi anemia. As discussed above, Fanconi anemia is an extremely genetically heterogeneous disorder due to autosomal recessive inheritance of homozygous mutations in genes, including BRCA1 and BRCA2 (for which autosomal dominant inheritance of heterozygous mutations are a cause of hereditary breast and ovarian cancer syndrome). Individuals with Fanconi anemia typically have a variety of birth defects, such as renal abnormalities and radial bone deformities, and an extremely elevated risk for developing acute myelogenous leukemia and squamous cell carcinoma of the head and neck. Cells from individuals with Fanconi anemia are sensitive to DNA alkylating agents (such as diepoxybutane and mitomycin C) and demonstrate chromosome breakage with unusual chromosomal tetrad formation during mitosis. Treatment of cultured patient cells with either of these agents forms the basis of a once commonly used (but now increasingly obsolete) clinical test for confirming the diagnosis of Fanconi anemia; however, other tests are available, including multigene panels employing next-generation DNA sequencing.
Ataxia telangiectasia. The defective gene in this autosomal recessive disorder, ATM, ordinarily recognizes broken chromosomes and participates in a cell cycle checkpoint to allow sufficient time for DNA repair to transpire. Affected individuals have a greatly elevated risk for hematopoietic and other malignancy. They also have a neurodegenerative syndrome characterized by an ataxic movement disorder. Telangiectasias are a cutaneous manifestation of this disease that are distributed on the sclerae and in a malar pattern across the face. Cells from individuals with ataxia telangiectasia may demonstrate increased chromosome breakage upon exposure to the DNA cross-linking agent bleomycin, which was used for diagnostic testing before the gene was identified and could be genetically tested for.
Xeroderma pigmentosum. This is a genetically heterogeneous autosomal recessive disorder, resulting from mutations in various transcription factors and nucleases. Individuals with xeroderma pigmentosum are at extreme risk for developing skin cancer upon exposure to sunlight due to a cellular defect in “excision repair” of ultraviolet light-induced thymine dimers.
Summary of cancer genetics
- Rare causes of cancer have enlightened the understanding of common tumors:
- Studies of RNA transforming viruses led to the discovery of proto-oncogenes that are somatically mutated (and dominantly activated) in many forms of cancer.
- Studies of cancer families have led to the identification of tumor suppressor genes, showing frequent inactivation (of both alleles) in sporadic cases of similar type.
- Cancer is, in general, a multistep pathway requiring the accumulation of mutations in several proto-oncogenes or tumor suppressor genes. This process can be accelerated if a DNA repair gene is inactivated.