
4 Other types of inheritance
Session Learning Objectives and a quick synopsis:
While most single gene disorders obey straightforward rules of inheritance, complicated loopholes in Mendel’s laws modify how some clinical traits are inherited: Genes encoded in the mitochondria are transmitted maternally. Uniparental gene expression for certain genes is due to imprinting, in which only one parental allele is expressed, or uniparental disomy, in which both alleles are derived from the same parent. New mutations give rise to the sudden appearance of a trait in a family and also account for germline and somatic mosaicism. The phenomenon of anticipation leads to worsening clinical severity or earlier age of onset for some disorders from one generation to the next due to mutations caused by unstable repetitive DNA sequences or shortening of the ends of chromosomes (telomeres).
SLO 1Describe features of mitochondrial inheritance
Mitochondrial DNA is inherited from maternal mitochondria (with rare exception), meaning that inheritance of disorders caused by mutations in mitochondrial genes is from mother to child.
SLO 2Distinguish maternal from paternal imprinting
A handful of autosomal genes are imprinted. Maternal imprinting means that the allele of a particular gene inherited from the mother is transcriptionally silent, and the paternally inherited allele is active. If a mutation in a maternally imprinted gene causes a dominant disorder, then that disorder cannot be inherited from the mother because the maternal allele is not expressed, anyway. A disease-causing mutation in such a gene can only be inherited from the father (who may or may not be affected, depending upon which of his parents he inherited the mutation from). Paternal imprinting is the opposite; the paternally inherited allele is silenced, and the maternally inherited allele is active. If a mutation in a paternally imprinted gene causes a dominant disorder, then that disorder cannot be inherited from the father because the paternal allele is not expressed, anyway. A disease-causing mutation in such a gene can only be inherited from the mother (who may or may not be affected, depending upon which of her parents she inherited the mutation from). An unaffected father who inherited a maternally imprinted disease-causing gene mutation from his mother has a 1/2 probability of transmitting the disorder to a child, regardless of the sex of the child. An unaffected mother who inherited a paternally imprinted disease-causing gene mutation from her father has a 1/2 probability of transmitting the disorder to a child, regardless of the sex of the child.
SLO 3Explain how uniparental disomy can give rise to an autosomal recessive or imprinted disorder
Uniparental disomy (also known as isodisomy) occurs when both alleles for a gene are inherited from the same parent. In other words, that particular allele is duplicated, and the allele from the opposite parent is absent. While several mechanisms can cause this to occur, typically it results from a chromosome nondisjunction event early during embryogenesis. In that case, both chromosomes are inherited from the same parent.
If an allele responsible for an autosomal recessive disorder resides on a chromosome duplicated due to isodisomy, then it will cause that disorder because the affected individual will be homozygous for the mutation.
If a gene affected by uniparental disomy is one that is ordinarily maternally imprinted and it is the maternal allele that is duplicated, then there will be no expression of that gene (because the paternal allele from which it is normally expressed is lost), resulting in disease. If a gene affected by uniparental disomy is one that is ordinarily paternally imprinted and it is the paternal allele that is duplicated, then there will be no expression of that gene (because the maternal allele from which it is normally expressed is lost, resulting in disease).
SLO 4Explain how new mutation accounts for seemingly sporadic occurrences of genetic disorders
Mutations must begin somewhere. When they occur for the first time in a gamete, then the conceptus is the first in the family to inherit that disease. The likelihood of this happening is inversely proportional to the severity of the disease. A disease that is lethal in childhood can only occur sporadically because those who have it do not live to reproductive age.
SLO 6Describe how lethal autosomal dominant mutations still give rise to disease
Some mutations are so deleterious that if all the cells of the body contained the mutation then the embryo could not survive. Nevertheless, a mutation can occur in a particular cell during embryogenesis, leading to a subset of tissue containing the cell with the mutation, thereby restricting disease manifestations to certain parts of the body.
SLO 7Describe genetic anticipation and explain how it alters patterns of inheritance
Genetic anticipation refers to worsening severity or earlier age of onset for a disease from one generation to the next. It is caused by mutations involving some genes containing an unstable repetitive element that intergenerationally increases in length, often in a parent of origin specific manner. Anticipation can also be caused by progressive shortening of telomeres, which are repetitive sequences at the ends of chromosomes, when there are mutations in genes encoding telomerase, which is responsible for maintaining telomere length, particularly in the germ cell compartment.
Main text
SLO 1Describe features of mitochondrial inheritance
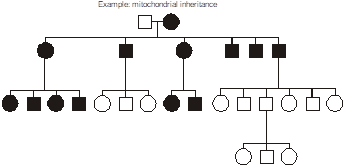 Maternally inherited transmission occurs when the defective gene is encoded in the mitochondrial genome. The mitochondria are organelles in the cytoplasm that perform aerobic energy metabolism. Because the cytoplasmic volume of an oocyte and their number of mitochondria is so vastly larger than for spermatozoa, mitochondria are inherited nearly exclusively from the oocyte during the formation of the zygote making them nearly exclusively maternal in origin. Mitochondria contain their own circular DNA genome plus RNA polymerases and protein translation apparatus including ribosomes and tRNA. That genome has only sixteen thousand base pairs and uses a genetic code that is slightly from the nuclear genome. Most children of an affected female are usually affected, to varying degrees.
Maternally inherited transmission occurs when the defective gene is encoded in the mitochondrial genome. The mitochondria are organelles in the cytoplasm that perform aerobic energy metabolism. Because the cytoplasmic volume of an oocyte and their number of mitochondria is so vastly larger than for spermatozoa, mitochondria are inherited nearly exclusively from the oocyte during the formation of the zygote making them nearly exclusively maternal in origin. Mitochondria contain their own circular DNA genome plus RNA polymerases and protein translation apparatus including ribosomes and tRNA. That genome has only sixteen thousand base pairs and uses a genetic code that is slightly from the nuclear genome. Most children of an affected female are usually affected, to varying degrees.
Disorders caused by mitochondrial gene mutations typically involve nerves and muscle. Examples include myoclonus epilepsy with ragged red fibers (MERRF); mitochondrial myopathy, encephalopathy, lactic-acidosis, and stroke-like episodes (MELAS), and Kearns-Sayre syndrome. Often they arise from point mutations, meaning that they result from substitution, deletion, or insertion of a single DNA base pair. These diseases tend to have some degree of clinical overlap, and the first two are pretty much as described by their lengthy names. In particular, ragged red skeletal muscle fibers, evident with a special stain, is a diagnostic feature of mitochondrial disorders.
Heteroplasmy vs. Homoplasmy – In mitochondrial inheritance, the disease is conventionally thought to be inherited exclusively from the mother, and usually all of an affected mother’s children also inherit the disorder. However, sometimes children can escape inheriting a mitochondrial disorder or can be affected to variable degrees, depending upon how the mutation is distributed within the heterogeneous population of hundreds of mitochondria inherited from the mother. Additionally, a non-penetrant mother may sometimes transmit disease to her children. Therefore, the proportion of mutant mitochondrial molecules contributes to the penetrance and expressivity of some mitochondrial disorders. This concept is referred to as “heteroplasmy.” The contrasting term, “homoplasmy,” refers to a cell containing a uniform population of mitochondrial DNA, either entirely wild type or entirely mutant; correspondingly, the child inherits a uniform population of mitochondrial genomes from the mother.
Not all the proteins responsible for mitochondrial function are encoded by the mitochondrial genome; some are products of ordinary, nuclear genes, and this has an interesting implication. A rare but intriguing class of mitochondrial disorders are the result of mutations in the mitochondrial genome, but the inheritance is “Mendelian.” In these unusual disorders, defects in nuclear genes encoding enzymes involved in mitochondrial DNA replication lead to frequent acquired mutations in the mitochondrial genome in many cells. These disorders typically present with neuromuscular features, similar to other mitochondrial disorders.
SLO 2Distinguish maternal from paternal imprinting
Normally, genes are equally well expressed from both parental alleles. But for a small number of genes, about 75 or so in humans, expression occurs only from either the maternal or paternal allele. They are said to be imprinted. For a subset of imprinted genes, only the maternal allele is expressed. For a different subset of imprinted genes only the paternal allele is expressed. For disease-causing mutations in imprinted genes, inheritance occurs only from one parent, because the gene is expressed only from either the maternally or paternally inherited allele. The most extreme examples of imprinting are the tumors, hydatidiform mole and ovarian teratoma. Hydatidiform mole results from fertilization of a polar body (an anucleate biproduct of female meiosis, more on this later) by an X-chromosome-containing sperm; the result is all placenta without an embryo. In contrast, ovarian teratoma results from an egg spontaneously becoming diploid and acting as if it has been fertilized; it gives rise to embryonic differentiation, but no placenta. Both are 46,XX and therefore chromosomally identical. The only difference between these two (and a normal female conceptus) involves the differential expression of just a handful of imprinted genes required for normal development.
With imprinting, gene expression occurs from only one parent’s allele (the maternal or the paternal, depending upon the gene). For all known imprinted genes, the particular gene and the particular parental allele that is expressed or silenced is constant from one person to the next. Disorders associated with imprinted genes can only be inherited from either the mother or the father, depending upon the gene causing the disorder.
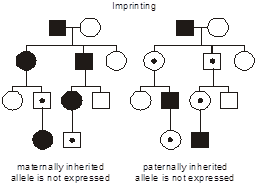 In the figure, consider two hypothetical, identically structured pedigrees. (We put a dot in the middle of the symbols for individuals who possess the mutant allele yet are unaffected.) The family on the left transmits a disease caused by a maternally imprinted gene where only the paternally inherited allele is expressed (i.e., transcribed). Consequently, if the mutation is inherited from the father, then only the mutant gene is expressed; since the normal, maternal allele is not expressed, then the phenotype appears. The reverse situation is considered in the pedigree on the right: here the disease results from mutation of a paternally imprinted gene where only the maternally inherited allele is expressed. Thus, when the mutation is inherited from the mother, there is no compensatory expression of the normal, paternal allele, and the phenotype emerges. Note that there is still a 1/2 probability that the mutant, vs. normal, allele will be transmitted at each meiosis, and so imprinting is superimposed on an autosomal dominant pattern of inheritance. Further note that it is immaterial whether the parent is affected or not—only the sex of the transmitting parent determines whether inheritance of the imprinted gene will cause the disease.
In the figure, consider two hypothetical, identically structured pedigrees. (We put a dot in the middle of the symbols for individuals who possess the mutant allele yet are unaffected.) The family on the left transmits a disease caused by a maternally imprinted gene where only the paternally inherited allele is expressed (i.e., transcribed). Consequently, if the mutation is inherited from the father, then only the mutant gene is expressed; since the normal, maternal allele is not expressed, then the phenotype appears. The reverse situation is considered in the pedigree on the right: here the disease results from mutation of a paternally imprinted gene where only the maternally inherited allele is expressed. Thus, when the mutation is inherited from the mother, there is no compensatory expression of the normal, paternal allele, and the phenotype emerges. Note that there is still a 1/2 probability that the mutant, vs. normal, allele will be transmitted at each meiosis, and so imprinting is superimposed on an autosomal dominant pattern of inheritance. Further note that it is immaterial whether the parent is affected or not—only the sex of the transmitting parent determines whether inheritance of the imprinted gene will cause the disease.
To reiterate, “paternal imprinting” specifically means that that the paternal allele for that gene is silenced and that only the maternal allele is expressed, and “maternal imprinting” means that the maternal allele for that gene is silenced and only the paternal allele is expressed.
Prader-Willi and Angelman syndrome are the best-known examples of diseases resulting from imprinting. And, they add another wrinkle to the complexity of imprinting; these two diseases result from oppositely imprinted genes in the same region of chromosome 15. Prader-Willi is inherited through mutations in the paternal homolog of this region of chromosome 15; Angelman syndrome results from inheritance of identical or different mutations in the maternal homolog of this chromosomal region.
In Prader-Willi and Angelman syndromes, an identical “chromosomal microdeletion” (also known as “copy number variant”) of several genes in a critical region of chromosome 15 can have a different disease outcome depending on which parent it is inherited from. Prader-Willi syndrome is clinically characterized in the neonatal period by failure to thrive, hypotonia, and mild to moderate intellectual disability. Later in childhood, these individuals have difficulty with satiety and have a notorious appetite. (One source for advice about this illness advises: “Refrigerators, cupboards, and garbage cans need to be locked.”) Consequently, individuals with Prader-Willi are quite obese. The oppositely imprinted counterpart of Prader-Willi syndrome is Angelman syndrome and consists of a somewhat small body habitus with severe intellectual disability and a marionette-like scissoring gait.
The molecular genetics of Prader-Willi and Angelman syndrome are complicated. Suffice it to say that there is an “imprinting center,” a small region within the imprinted domain on chromosome 15 that exerts a more regional effect on gene expression from this portion of the chromosome. The imprinting mechanism relies on alternatively spliced short transcripts and site-specific DNA methylation in this region. There are at least two nearby and oppositely imprinted genes, SNRPN, encoding a protein that functions in pre-mRNA processing and may contribute to tissue-specific alternative splicing, and UBE3A, an E3 ubiquitin ligase regulating proteasome-mediated proteolytic degradation of other proteins. Mutations in SNRPN appear sufficient to cause Prader-Willi syndrome, when inherited from the father. Mutations in UBE3A cause Angelman syndrome, when inherited from the mother. For some patients, other mutations in this region may contribute. A deletion large enough to remove both SNRPN and UBE3A can cause both Prader-Willi and Angelman syndrome to appear in the same family, depending upon the sex of the transmitting parent.
The biological role of imprinting remains unclear. Most of the handful of genes that are known to be imprinted are generally responsible for growth effects in fetal life. Some have argued that this represents a battle of the sexes at the molecular and cellular level, in which the father imprints these genes in an “on” position to insure that they are expressed in the conception that he has fathered so that he will have big, presumably healthier, kids, whereas the mother imprints these genes in the “off” position, so that her fetus will not grow too large and endanger her own well-being.
SLO 3Explain how uniparental disomy can give rise to an autosomal recessive or imprinted disorder
Uniparental disomy occurs when both copies of a particular chromosome (or even only part of a chromosome) are inherited from just one parent, with no contribution from the other parent.
Given that the chromosome 15 region responsible for Prader-Willi and Angelman syndromes is imprinted, it is not surprising that uniparental disomy can also be a mechanism for these two disorders. In particular, maternal uniparental disomy for chromosome 15, in which there is no contribution of essential genes only expressed from paternal alleles, is one cause of Prader-Willi syndrome.
Uniparental disomy does not require imprinting in order for it to be a genetic cause of disease. In fact, it was first discovered in a situation where an autosomal recessive disease was found in a child where only one of her parents was a carrier for the mutant allele. In that case, a child had cystic fibrosis. Only her mother was a heterozygous carrier of a mutant allele for the CFTR gene on chromosome 7, and her father was homozygous wild type. Molecular investigations revealed that both of the child’s copies of chromosome 7 were of maternal origin, thereby conferring upon the child two mutant alleles for CFTR. This child also had excessively short stature. Presumably, uniparental disomy also had an effect on one or more other genes on chromosome 7 capable of governing skeletal growth, either through duplicating a variant allele that behaved recessively or through an imprinting effect.
The frequency with which uniparental disomy occurs is difficult to estimate, since if disease-causing alleles of the disomic genes are not involved, or the disomic region is not imprinted, then its occurrence will likely be clinically inconsequential and go undetected. Probably the most common mechanism for generating uniparental disomy is the “rescue” of a trisomic conception. We will revisit this topic when we discuss chromosomal disorders.
SLO 4Explain how new mutation accounts for seemingly sporadic occurrences of genetic disorders
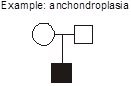 Example: achondroplasia. What are the explanations for this pedigree in which two unaffected parents give birth to a child with achondroplasia? Achondroplasia is an autosomal dominant disorder of short-limbed short stature resulting from a single base pair mutation leading to amino acid substitution in the fibroblast growth factor receptor 3 (FGFR3) gene. In this family, there are two normally statured and unaffected parents who have a child with achondroplasia. We can exclude incomplete penetrance in the parents because achondroplasia is known to be completely penetrant. We should consider three possible explanations. First, non-paternity (or perhaps less commonly, non-maternity) is always a possibility. A second possible explanation that we will discuss shortly is germline mosaicism. Germline mosaicism has been documented to occur for achondroplasia, but it is a rare phenomenon. At this point we have no reason to invoke it as a consideration for this pregnancy, but if this couple should have two children with achondroplasia then this would be the most probable explanation. The third possibility, however, given the current pedigree with just one affected child, is the most probable, and that is the child represents a new mutation (also known as “de novo” mutation). In other words, the mutation occurred for the first time in this family’s history, in the affected child.
Example: achondroplasia. What are the explanations for this pedigree in which two unaffected parents give birth to a child with achondroplasia? Achondroplasia is an autosomal dominant disorder of short-limbed short stature resulting from a single base pair mutation leading to amino acid substitution in the fibroblast growth factor receptor 3 (FGFR3) gene. In this family, there are two normally statured and unaffected parents who have a child with achondroplasia. We can exclude incomplete penetrance in the parents because achondroplasia is known to be completely penetrant. We should consider three possible explanations. First, non-paternity (or perhaps less commonly, non-maternity) is always a possibility. A second possible explanation that we will discuss shortly is germline mosaicism. Germline mosaicism has been documented to occur for achondroplasia, but it is a rare phenomenon. At this point we have no reason to invoke it as a consideration for this pregnancy, but if this couple should have two children with achondroplasia then this would be the most probable explanation. The third possibility, however, given the current pedigree with just one affected child, is the most probable, and that is the child represents a new mutation (also known as “de novo” mutation). In other words, the mutation occurred for the first time in this family’s history, in the affected child.
Whenever a child with a highly penetrant autosomal dominant disease is born to two unaffected parents, then the possibility of new mutation should be considered. When a second affected child is born to the same couple, then germline mosaicism becomes a stronger possibility.
Typically, the mutation would occur in a gamete (egg or sperm) that gave rise to that individual. The mutation could also occur in the zygote or very early post-zygotically such that it was present in the majority of the cells of the embryo. If it arose a few cell divisions later in the embryo, then it would be distributed in a mosaic fashion in the body. New mutations represent about a third of all cases of achondroplasia, and, therefore, a family like this is not uncommon. It turns out that for achondroplasia, the overwhelming majority of the new mutations occur on the paternal chromosome and appear to be associated with advanced paternal age.
It is estimated that every child has about 100 new genomic DNA sequence changes, not found in either parent. The majority occur on paternally inherited chromosomes. Most are inconsequential because functional gene sequences constitute only a small portion of the genome and, for those few new mutations that do reside within a gene, some are not pathogenic or behave recessively, in which case a heterozygote would be unaffected.
In general, advanced paternal age is associated with an increased risk for new mutations in a single gene, Mendelian disorder, while, as we shall discuss later, advanced maternal age is associated with a risk for chromosomal disorders resulting from meiotic non-disjunction. The reason for this is because mutations are often linked to DNA replication, occurring during cell division. The formation of sperm is ongoing throughout life. However, the production of oocytes requires no further mitotic cell division beyond early fetal life.
For serious autosomal dominant illnesses, new mutations account for 100% of cases. Why should it be so high? The answer is because the genetic “fitness” (successful reproduction) is reduced. Individuals affected with serious disease either die before reproductive age or are otherwise too sick to reproduce. To account for a reasonable assumption of a constant disease incidence in the population over time, the alleles lost to lethal events must be replaced by new mutations.
For example, heterozygous mutations in the gene, ELANE, encoding the serine protease neutrophil elastase are the most common cause of severe congenital neutropenia. Neutropenic individuals have a low number of circulating neutrophils (phagocytic white blood cells) and succumb to opportunistic infections or are, at the least, otherwise too sick to become parents. Consequently, nearly all mutations in ELANE responsible for severe congenital neutropenia arise newly, and there is seldom a family history of neutropenia.
It is reasonable to suppose that the incidence of a genetic disease should remain constant in a population over time. Since lethal alleles will be lost in the population on account of poor genetic fitness, it stands to reason then that the rate at which new mutation generates new disease alleles will equal the rate at which existing alleles are lost from the population. Based on this premise, one-third of all those affected with a severe sex-linked recessive disease will represent new mutations, without a prior family history of that disorder. Let’s look at the situation in sex-linked recessive diseases that are lethal, where fitness is zero. All the alleles in an affected male are lost from the population in the next generation and must have been replaced with new mutations if the incidence of the disease remains constant over time. Since one-third of all X chromosomes in the population collectively reside in males, then one-third of all lethal alleles should be new mutations. In fact, this is observed to be true for Duchenne muscular dystrophy and other severe sex-linked recessive disorders.
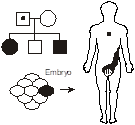 Germline mosaicism occurs when the original mutation arose post-zygotically in the affected parent such that only a fraction of the parent’s cells contains the mutation. In particular, the germ cells giving rise to the gametes do contain the mutation, but the mutation is in such minority in the rest of the tissues of the body so as to not produce a recognizable phenotype. It explains the recurrence of a highly penetrant autosomal dominant disease in a family with two unaffected parents. An example occurs with osteogenesis imperfecta, an autosomal dominant disorder of collagen producing brittle bones prone to recurrent breakage. A particularly striking case of germline mosaicism was found when a male without osteogenesis imperfecta fathered two children with the disease by two different females. The collagen mutation could be molecularly identified and was identical in both children but appeared absent from DNA extracted from peripheral white blood cells in the father. However, individual hairs were plucked from the father’s head and then subjected to mutational analysis. As it turned out, some of the hair shafts had the mutation and others didn’t—demonstrating mosaicism down to the level of the hair follicle.
Germline mosaicism occurs when the original mutation arose post-zygotically in the affected parent such that only a fraction of the parent’s cells contains the mutation. In particular, the germ cells giving rise to the gametes do contain the mutation, but the mutation is in such minority in the rest of the tissues of the body so as to not produce a recognizable phenotype. It explains the recurrence of a highly penetrant autosomal dominant disease in a family with two unaffected parents. An example occurs with osteogenesis imperfecta, an autosomal dominant disorder of collagen producing brittle bones prone to recurrent breakage. A particularly striking case of germline mosaicism was found when a male without osteogenesis imperfecta fathered two children with the disease by two different females. The collagen mutation could be molecularly identified and was identical in both children but appeared absent from DNA extracted from peripheral white blood cells in the father. However, individual hairs were plucked from the father’s head and then subjected to mutational analysis. As it turned out, some of the hair shafts had the mutation and others didn’t—demonstrating mosaicism down to the level of the hair follicle.
Germline mosaicism should be considered as a possible explanation for a couple in which neither parent is affected yet has more than one child with a highly penetrant autosomal dominant illness.
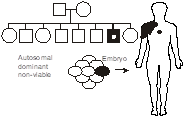 SLO 6Describe how lethal autosomal dominant mutations still give rise to disease
SLO 6Describe how lethal autosomal dominant mutations still give rise to disease
There are mutations in some genes that would result in a non-viable pregnancy. Consequently, they are only seen in a mosaic state in affected individuals, in which the mutation is confined to a small patch of tissue. An example is McCune-Albright syndrome. McCune-Albright syndrome is virtually never inherited, as would be expected with such a disease, and results from activating mutations in the alpha subunit of the stimulatory G protein Gs that regulates intracellular cAMP signaling. Clinically, McCune-Albright is characterized by multiple endocrine tumors comprising somatic foci of constitutively activated cells.
SLO 7Describe genetic anticipation and explain how it alters patterns of inheritance
Anticipation – the clinical observation that an inherited disease displays increasing severity and/or an earlier age of onset with each subsequent generation.
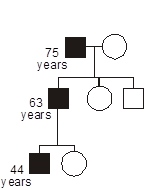 Some heritable disorders demonstrate genetic anticipation, a declining age of onset or worsening severity with each passing generation. Anticipation is a feature of disorders where the gene mutation consists of an unstable short tandem repeat sequence, nearly all of which are developmental or degenerative neurological diseases. Anticipation can also occur with disorders related to preservation of telomere length.
Some heritable disorders demonstrate genetic anticipation, a declining age of onset or worsening severity with each passing generation. Anticipation is a feature of disorders where the gene mutation consists of an unstable short tandem repeat sequence, nearly all of which are developmental or degenerative neurological diseases. Anticipation can also occur with disorders related to preservation of telomere length.
Fragile X syndrome. Fragile X syndrome is among the most common causes of intellectual disability for both males and females. Affected individuals may also have characteristic dysmorphic features, including long facies (fancy medical term for face) with prominence of the ears, forehead, and jaw. Most postpubertal males with fragile X have enlarged testes (“macro-orchidism”). The disease takes its name from the fact that most affected individuals have a “fragile site” demonstrable at the terminus of the long arm of the X chromosome.
Chromosome fragile sites are gaps or constrictions that are vulnerable to breakage in otherwise condensed chromatin on metaphase chromosomes. They become apparent only during in vitro cell culture under certain conditions. The genomic DNA sequence at fragile sites tends to consist of long runs of di or triplet nucleotide repeats. Fragile sites are considered part of normal chromosome structure, and numerous fragile sites are normally present at defined positions throughout the genome in most people. A few fragile sites, however, are rare and are associated with disease.
The fragile X site, for example, looks like a small bit of the end of the chromosome has broken off and is hanging on thread-like to the rest of the chromosome. While the fragile X site may be vulnerable to breakage, breakage of the chromosome is not what causes fragile X syndrome. Instead, it is caused by aberrant expression of a particular gene due to changes in the length of a triplet repeat sequence that is contained within that gene; the longer repeat also leads to appearance of the fragile site in laboratory-cultured cells.
Like most hereditary illnesses resulting from mutation of a gene on the X chromosome, fragile X is inherited as a sex-linked recessive disorder. Males inherit the disease from a carrier mother. The penetrance for females is rather high, however, and results from skewing of X chromosome inactivation in affected females.
The inheritance of fragile X is not, however, quite as straightforward as it is for other sex-linked recessive diseases. Consistent with anticipation, the likelihood of being affected with fragile X is dependent on the position of the individual in the pedigree. Individuals appearing in a generation subsequent to one in which somebody is already known to have the disease are at a higher risk for having affected children.
Fragile X results from mutations in a gene, FMR1, at the location of the fragile site on the X chromosome. The protein product of the gene binds RNA and appears to function in the nuclear export of RNA. The mutation involves “expansion” of a tandemly repeated CGG DNA sequence. In normal individuals there are about 8 to 50 copies of a CGG trinucleotide repeat in the 5’ untranslated region (5’-UTR) containing the promoter regulating FMR1 expression. Individuals with fragile X syndrome have a mutation comprised of about 200 to 1,000 copies of this repetitive trinucleotide sequence. There is a third category of individual known as “premutation carriers,” who are unaffected with fragile X, but who are at risk for having children or later descendants with fragile X, who have a repeat length that’s somewhere in-between. Affected individuals with the “full mutation” have greatly reduced transcriptional expression of the fragile X gene from the chromosome containing the expanded repeat. The multiple CpG sequences resulting from repeat expansion provide more targets for DNA methylation. As is usual with cytosine methylation in the promoter region of a gene, transcription is switched off, thereby resulting in loss of expression of that allele. There are extremely rare individuals who have fragile X syndrome resulting, not from expansion of CGG repeats, but rather as a result of an inactivating point mutation in the coding sequence of the gene, confirming that fragile X syndrome is caused by loss of expression of FMR1.
Genetic testing, typically using the polymerase chain reaction (PCR) or other methods to measure the length of the CGG repeat tract, is available for the fragile X syndrome and has replaced the much less sensitive and older approach based on cell culture to demonstrate extrusion of the fragile site on the X chromosome.
Although complicated, the inheritance of fragile X syndrome is predictable.
The important concept to appreciate is that once CGG repeat sequences (or other repeat sequences associated with other diseases) have expanded to a certain length, they become unstable during meiosis and are at risk of expanding to an even greater length in each subsequent generation. There is some threshold number of repeats that, once crossed, turns a premutation into a full mutation capable of causing the disease. For most disorders caused by expansion of short repeat sequences, meiotic expansion of the repeat tends to occur only when inherited from the mother (as is the case for fragile X) or from the father, depending upon the disorder.
Expansion of the fragile X CGG triplet repeat from a normal length to the premutation length almost never happens. The premutation appears to have been inherited from just a few common ancestral founders in the general population.
On the other hand, expansion of a premutation-length repeat to a full mutation is rather common but can only occur through a female meiosis. For unexplained reasons, for each particular disease caused by a repeat expansion, the expansion is characteristically more likely to occur when inherited from a parent of one sex or the other.
In general, for fragile X syndrome and other disorders caused by unstable short repeats, the risk that a premutation will expand to a full mutation is dependent upon the length of the premutation repeat in the parent from whom it is inherited. The greater the length of the premutation, then the more likely it is to expand during meiosis. In general, however, for fragile X syndrome, we can assume there to be about an 80% probability that a premutation repeat will expand to a full mutation length during female meiosis. However, when the premutation repeat length reaches 90 or more triplets, then expansion to a full mutation happens with near certainty during female meiosis.
The penetrance of fragile X in males with the full mutation is 100%. In females with the full mutation, the penetrance is about 50% (as a result of skewed Lyonization).
Premutation carriers exhibit different symptoms than those with a full mutation. Female fragile X premutation carriers tend to have premature ovarian failure (i.e., early onset of menopause), and male fragile X premutation carriers tend to develop tremors and ataxia (poorly coordinated movement) with advancing age, a disorder known as fragile X–associated tremor/ataxia syndrome (FXTAS). Paradoxically, whereas the full CGG repeat mutation tends to extinguish expression of the FMR1 gene, the premutation tends to elevate its expression.
Huntington disease. Huntington disease is an autosomal dominant, adult-onset, severe neurodegenerative disorder. It is characterized in the initial stages by mild cognitive symptoms with progressive choreiform (involuntary dance-like) movement disorder. At the neuroanatomic level, there is degeneration of the caudate and putamen in the basal ganglia of the brain. The responsible gene, HTT, is expressed ubiquitously throughout the brain. Molecular pathogenesis of the disorder remains a subject of investigation, but the protein encoded by HTT plays a role in vesicle trafficking and endocytosis.
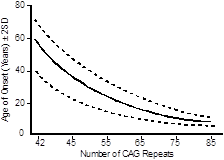 The mutation in every individual with Huntington disease is a variably sized tandem expansion of the DNA triplet repeat sequence CAG, contained in the protein-coding sequence of the gene, and encoding the amino acid glutamine. Individuals without Huntington disease normally have fewer than 35 CAG repeats, although the repeat length is polymorphic among the normal population. Individuals with 40 or more CAG repeats usually develop Huntington disease. The protein encoded by HTT from individuals with a CAG repeat tract expansion will then have an elongated polyglutamine tract within the polypeptide.
The mutation in every individual with Huntington disease is a variably sized tandem expansion of the DNA triplet repeat sequence CAG, contained in the protein-coding sequence of the gene, and encoding the amino acid glutamine. Individuals without Huntington disease normally have fewer than 35 CAG repeats, although the repeat length is polymorphic among the normal population. Individuals with 40 or more CAG repeats usually develop Huntington disease. The protein encoded by HTT from individuals with a CAG repeat tract expansion will then have an elongated polyglutamine tract within the polypeptide.
The age of onset of the disease can be largely explained by the number of repeats an individual possesses—the greater the number of repeats, then the earlier the age of onset. Just as with fragile X syndrome, once the number of repeats passes a critical threshold, then the repeat tract becomes unstable and is at risk for further increases in length with every subsequent meiosis. Unlike the case for fragile X syndrome, meiotic repeat expansion, especially large expansions, occurs preferentially during paternal meiosis.
For most diseases where the mutation is caused by an unstable short DNA repeat there is a strong inverse correlation between the length of the repeat and the age of onset or severity of the disease.
 Presymptomatic diagnostic testing can determine if one has inherited a disease-causing mutation before symptoms are apparent. The test for Huntington disease is simple and consists of a PCR assay to determine the length of the CAG repeat tract. Ethical issues relating to the use of the test are not so simple, however. It is recommended that individuals at risk for inheriting Huntington disease who undergo molecular genetic testing for presymptomatic diagnosis receive appropriate pre-test genetic counseling and post-test psychological support. Similar recommendations apply to presymptomatic diagnostic testing for any disorder with potentially profound impacts on health and longevity.
Presymptomatic diagnostic testing can determine if one has inherited a disease-causing mutation before symptoms are apparent. The test for Huntington disease is simple and consists of a PCR assay to determine the length of the CAG repeat tract. Ethical issues relating to the use of the test are not so simple, however. It is recommended that individuals at risk for inheriting Huntington disease who undergo molecular genetic testing for presymptomatic diagnosis receive appropriate pre-test genetic counseling and post-test psychological support. Similar recommendations apply to presymptomatic diagnostic testing for any disorder with potentially profound impacts on health and longevity.
Other diseases caused by unstable short tandem repeats. There are several human genetic disorders caused by short tandem DNA repeat expansion. While they all play by a unique set of complicated rules, their inheritance patterns are for the most part predictable, there is a set of common clinical features, and genetic testing can precisely define risk.
Characteristics of diseases caused by short tandem repeat expansion
- The short tandem DNA repeats, depending upon the disease gene in which they reside, can be located in upstream regulatory regions, within an intron, within untranslated regions of mRNA or in the coding sequence where they are translated (in particular for CAG repeats, translated into runs of the amino acid glutamine).
- Disease-associated alleles can exist in “pre-mutation” and “full-mutation” states, with the two states being differentiated by the length of the expanded tract of repeats.
- The repeat length at most disease loci is variable in the normal population. Expansion from a normal length repeat to a premutation repeat happens only infrequently, but once a premutation has occurred, there is a high likelihood of further meiotic expansion to a full mutation state.
- In general, expansion from premutation to full mutation shows a parent-specific effect (either maternal or paternal) for each particular disorder. The expansion occurs during meiosis.
- The length of the repeat tract inversely correlates with age of onset of the disease and accounts for anticipation, as revealed by an earlier age of onset or worsening severity with consecutive generations.
- At the molecular level, the repeats can be a site of epigenetic modification, such as promoter hypermethylation, and thereby lead to reduced or increased production of mRNA transcript or, if contained in a protein-coding sequence, altered proteins.
- Depending upon the particular gene and its associated disorder, pathogenesis may be caused by too much or too little of the normal gene product, a toxic mRNA transcript that can aggregate or sequester RNA-binding proteins, or a toxic protein capable of aggregating.
Anticipation with disorders of telomeres. Finally, another completely different mechanism for genetic anticipation occurs with disorders involving replication of telomeres, the structures at the ends of chromosomes. Due to the fact that DNA synthesis precedes only in the 5’ to 3’ direction, the ends of chromosomes represent a particular problem for DNA replication, and a special complex of proteins along with an RNA-priming template comprising telomerase and associated factors is required for maintenance of telomere length in stem cells, including germ cells. Mutations in any number of genes, including TERT, which encodes the protein component of telomerase, and TERC, which produces a non-protein-coding RNA, can cause the disorder dyskeratosis congenita. For most somatic cells, including somatic stem cells, there is progressive shortening of telomeres with each cell division. When telomeres become too short, cells can no longer divide. Telomere length therefore dictates the replicative lifespan of cells. In dyskeratosis congenita, telomere length in both somatic cells, as well as germ cells giving rise to sperm or eggs, is not maintained. Consequently, telomeres shorten with each passing generation in these disorders, resulting in short telomeres in all cells of a person inheriting the disorder. Cells with reduced telomere length cannot undergo as many cell divisions. The most rapidly dividing populations of cells are the most severely affected, including hair, skin, nails, bone marrow, and epithelial cells lining the lungs, leading to premature aging, aplastic anemia and bone marrow failure, and fibrotic lung disease, respectively. Moreover, the telomere length continues to shorten between generations, thereby leading to genetic anticipation with intergenerational worsening of symptoms occurring at progressively earlier ages of onset.