
3 Single gene inheritance patterns
Learning Objectives for these two sessions and a quick synopsis:
The overall goals are to distinguish the patterns of different forms of Mendelian inheritance (autosomal dominant, autosomal recessive, and sex-linked recessive inheritance) and identify concepts common to all forms of Mendelian inheritance.
Based on the sex and generation of the affected individuals in a family, the goal is to predict the most likely form of inheritance, which, in turn, can help narrow the differential diagnosis for clinical signs and symptoms of a particular disorder.
SLO 2Describe the expected pattern of inheritance within a family for an autosomal dominant disorder
Fully penetrant autosomal dominant disorders are passed from parent to child with a 1/2 probability, and do not depend upon the sex of the parent or child. Consequently, multiple generations within a family are typically affected. There are two copies of each autosomal gene; autosomal dominant inheritance requires that only one parentally contributed allele be mutated to produce a phenotype.
SLO 3Explain penetrance and situations where incomplete penetrance might be expected
Penetrance refers to whether the inherited allele produces a phenotype or not. Factors resulting in incomplete penetrance include age, sex, environment, and sometimes other inherited factors.
Bayes theorem is a mathematical concept for incorporating educated guesses and anecdotal observations into probability calculations. It is important for all of medicine, not just genetics, and informs our approach for everything ranging from ordering the right test to formulating a differential diagnosis. Although its mathematical formulation is challenging, it can be applied conceptually.
SLO 5Explain variable expressivity and what accounts for it
Variable expressivity is the concept that different people can have different manifestations of the same genetic disorder. The same factors contributing to incomplete penetrance help explain this phenomenon, but with the additional consideration that when comparing people from different families, there may be different disease alleles that differ between families.
Autosomal recessive inheritance typically requires that each parent be an unaffected carrier for the disease. Consequently, the disease is often restricted to just one generation in a family. When each parent is a carrier, fully penetrant autosomal recessive disorders are passed from parents to a child with a 1/4 probability and do not depend upon the sex of the child. It occurs more commonly in families in which there has been consanguinity (inbreeding) or whose ancestry descends from a small founding population. There are two copies of each autosomal gene; autosomal recessive inheritance requires that both parentally contributed alleles be mutated to produce a phenotype.
The frequency of disease alleles in a population can be used to determine the incidence of disease in a population. This information is important for providing genetic counseling. It also explains why the beneficial effects for unaffected heterozygous carriers of certain recessive disease alleles, who outnumber those who are affected, can lead to their persistence in a population despite their potential for producing disease.
Genes responsible for sex-linked recessive disorders reside on the X-chromosome, which is involved in biological sex determination. Consequently, penetrance is a function of sex. Males, with only copy of an X-chromosome gene, are typically affected when they inherit a mutant allele from an unaffected carrier mother, who possesses one disease-associated and one normal allele.
Main text
This learning objective really encompasses all of this chapter. Phenotypes may be inherited through one of several Mendelian patterns. We consider each in turn and introduce related concepts along with each form of inheritance. In general, multigenerational inheritance involving either sex suggests autosomal dominant inheritance. One mutant copy of the gene is sufficient to cause disease with an autosomal dominant disorder. Multiple affected individuals within a generation suggest autosomal recessive inheritance. In contrast to autosomal dominant disorders, one wild type copy of the gene is sufficient to prevent occurrence of an autosomal recessive disorder. Both copies of an autosomal gene must be mutated to cause disease with this form of inheritance. Suspicion for autosomal recessive inheritance should be heightened when there is inbreeding (consanguinity) or in populations descended from a small number of ancestors. Sex-linked recessive disorders usually affect males and are inherited from carrier mothers.
SLO 2Describe the expected pattern of inheritance within a family for an autosomal dominant disorder
Autosomal dominant inheritance. In autosomal dominant inheritance, the responsible gene is on an “autosome,” a chromosome that is not the X or Y sex chromosome. The mutation acts dominantly in that the normal allele is insufficient to compensate for the mutant allele. Heterozygotes with one copy of the disease allele and one normal allele are thus affected. Since one mutant allele has to overcome the effects of a normal allele simultaneously present within the cell, the molecular or cellular effect of the mutation is either “dominant negative,” where the mutant gene product adversely affects the wild type gene product, or “toxic gain of function” where the mutant gene product gains a new and damaging property, such as the mutant protein misfolds and “gums up” (to use non-technical jargon) the inner workings of the cell, or occasionally “haploinsufficiency” where half the level of expression from just the normal allele is not sufficient for normal gene activity.
Note that homozygotes for the disease allele are generally rare. For some diseases they are much more severely affected (to the extent that they may even be considered to have a different disorder), while for others there is no difference in phenotype with the heterozygous state. In general, you don’t need to worry about homozygotes for the diseased allele in autosomal dominant disorders, because of their rarity. “Codominance” refers to traits where homozygous wild type, heterozygotes, and homozygous mutant genotypes exhibit three distinct phenotypes, with the heterozygotes exhibiting severity somewhere in-between homozygous wild type or mutant. It can also be a feature of traits, such as blood types, that are not typically associated with disease.
Characteristics of autosomal dominant inheritance
- The phenotype appears in every generation, each affected person having an affected parent (except with reduced penetrance, new mutation, germline mosaicism, or anticipation).
- Each child of an affected parent has a 50% risk of inheriting the trait.
- Unaffected family members do not transmit the phenotype to their children (except with reduced penetrance, new mutation, germline mosaicism, or anticipation).
- Males and females are equally likely to transmit the trait, to children of either sex. In particular, there is male-to-male transmission (in contrast to sex-linked recessive inheritance).
- New mutations are relatively common, sometimes accounting for up to half or more of all patients, depending on the fitness of the trait.
 Example: Marfan syndrome. Marfan syndrome is a hereditary disease affecting the skeleton, eyes, and cardiovascular system. Skeletal manifestations are comprised of disproportionately long extremities including long fingers and toes (spider-like “arachnodactyly”), sternal chest deformity (“pectus excavatum” or simply “pectus”), and lateral curvature of the spine (“scoliosis”). Ophthalmologic abnormalities consist of near-sightedness (“myopia”) and lens dislocation (“ectopia lentis”). The major cardiovascular abnormality is a risk for aortic aneurysm and dissection. Mutations in the fibrillin (FBN1) gene on chromosome 15 are the most common cause of Marfan syndrome. Note that gene names are italicized; human gene names are written in all capital letters, whereas for other organisms, only the first letter is capitalized.
Example: Marfan syndrome. Marfan syndrome is a hereditary disease affecting the skeleton, eyes, and cardiovascular system. Skeletal manifestations are comprised of disproportionately long extremities including long fingers and toes (spider-like “arachnodactyly”), sternal chest deformity (“pectus excavatum” or simply “pectus”), and lateral curvature of the spine (“scoliosis”). Ophthalmologic abnormalities consist of near-sightedness (“myopia”) and lens dislocation (“ectopia lentis”). The major cardiovascular abnormality is a risk for aortic aneurysm and dissection. Mutations in the fibrillin (FBN1) gene on chromosome 15 are the most common cause of Marfan syndrome. Note that gene names are italicized; human gene names are written in all capital letters, whereas for other organisms, only the first letter is capitalized.
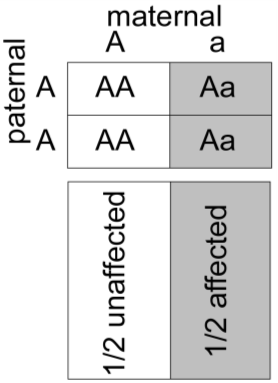
In this pedigree, every affected individual has a 1/2 probability of transmitting Marfan syndrome to each of their offspring. Thus, the chance that the fetus will inherit Marfan syndrome from the affected mother is 1/2.
One useful tool for illustrating Mendelian inheritance is the “Punnett” square. Here it can be seen that the affected mother’s genotype is A/a, where “a” represents the mutant allele for the gene causing Marfan syndrome and “A” represents a “normal” or non-disease-causing allele. We put the maternal genotype horizontally on top of the Punnett square and note that during maternal meiosis there is an equal (and hence 1/2) probability of transmitting either the a or A allele to the oocyte. We put the father’s genotype vertically on the side of the Punnett square. His genotype is denoted as A/A, since it is inferred that because he is unaffected he has two normal alleles of the fibrillin gene. Again, during paternal meiosis the chance of segregating either normal allele to the spermatozoa is equal and is 1/2. Since paternal and maternal meiosis are independent events, then we just multiply the individual probabilities to determine the probability that both will happen. From the Punnett square, then, we can see that there are four possible outcomes, each with probability of 1/2 × 1/2 = 1/4. The probabilities for all of the possible outcomes sums to one, since we know that one of these must actually happen. Two of the outcomes result in a conception that inherits Marfan syndrome with the genotype A/a, whereas two of the outcomes yield a conception that does not inherit Marfan syndrome (A/A). The probability that the conception will have inherited Marfan syndrome is therefore the number of the squares in the diagram that yield genotype A/a (two) divided by the total number of possible outcomes (four), which is 1/2. The probability that the conception will not have inherited Marfan syndrome is similarly the number of squares in the diagram that produce the genotype A/A (two squares), divided by the total number (four squares) or 1/2. Again, the sum of these mutually exclusive events is one.
SLO 3Explain penetrance and situations where incomplete penetrance might be expected
Penetrance vs. Expressivity. The phenotype may not appear in all individuals or, if it is present, may not be the same in different individuals. The former concept refers to “penetrance,” while the latter relates to “expressivity.”
Penetrance – Whether or not a mutation in a gene has any phenotypic expression at all. In contrast to expressivity, severity is not taken into account with the concept of penetrance, which is kind of an “all or none” description.
Example of incomplete penetrance: Factor V Leiden deficiency. Factor V is a component of the blood-clotting cascade. The Leiden mutation (named after the city in the Netherlands where it was discovered) is an amino acid missense substitution that impairs the function of blood clotting factor V, leading to resistance to activated protein C. Heterozygotes for the factor V Leiden allele develop a hypercoagulable state that leads to a risk for developing venous blood clots. As many as 40% of individuals of European-American ancestry presenting with deep venous thrombosis (DVT) may be factor V Leiden heterozygotes. Because the factor V Leiden allele confers hypercoagulability in the heterozygous state, it is inherited in an autosomal dominant fashion. The clinical phenotype, DVT, will manifest over the lifetime of only about 10% of heterozygotes, however. We call this phenomenon incomplete penetrance, in that not everyone who inherits the mutation will have clinical manifestations of the disease.
Can you think of reasons for incomplete penetrance? Other thrombosis risk factors include sedentary activity (such as airliner travel or a postoperative patient confined to bed), pregnancy, the presence of a central venous catheter commonly used for chemotherapy or long-term antibiotic administration, oral contraceptive use, cancer, and other genetic factors contributing to hypercoagulability. Thus, it is a combination of other genes and environmental factors differing between individuals that accounts for incomplete penetrance.
Example of sex-dependent penetrance: BRCA2-associated hereditary breast and ovarian cancer. BRCA2 is one gene responsible for hereditary breast and ovarian cancer. Males who belong to a BRCA2 breast cancer family can also develop breast cancer, albeit infrequently compared to females. With BRCA2 mutations, the penetrance for breast cancer is greatly reduced in males, an example of sex-dependent penetrance.
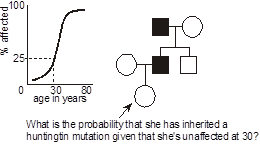 Example of age-dependent penetrance: Huntington disease. Huntington disease is an autosomal dominant neurodegenerative disease characterized by a dance-like movement disorder known as “chorea.” Anatomically, there is focal degeneration of the caudate nucleus in the brain. Mutations in the gene, HTT, are responsible for the disease. The mutations are always expansions of polyglutamine-encoding CAG trinucleotide repeat tracts (more on this later). The disease demonstrates age-dependent penetrance, in that, unlike some illnesses where the phenotype is present at birth, clinical manifestations, and hence diagnosis, rarely occur before adulthood. In this pedigree, we wish to calculate the probability that a woman who is presently 30 years-old inherited Huntington disease from her affected father, knowing that at this age she is asymptomatic. From published studies, we have available data correlating the age of onset of symptoms and signs of Huntington disease in individuals heterozygous for mutations of a certain-sized polyglutamine repeat. How do we go about applying this data toward the risk calculation posed in this example pedigree? To do so we need to discuss another aspect of probability, known as Bayes’ theorem.
Example of age-dependent penetrance: Huntington disease. Huntington disease is an autosomal dominant neurodegenerative disease characterized by a dance-like movement disorder known as “chorea.” Anatomically, there is focal degeneration of the caudate nucleus in the brain. Mutations in the gene, HTT, are responsible for the disease. The mutations are always expansions of polyglutamine-encoding CAG trinucleotide repeat tracts (more on this later). The disease demonstrates age-dependent penetrance, in that, unlike some illnesses where the phenotype is present at birth, clinical manifestations, and hence diagnosis, rarely occur before adulthood. In this pedigree, we wish to calculate the probability that a woman who is presently 30 years-old inherited Huntington disease from her affected father, knowing that at this age she is asymptomatic. From published studies, we have available data correlating the age of onset of symptoms and signs of Huntington disease in individuals heterozygous for mutations of a certain-sized polyglutamine repeat. How do we go about applying this data toward the risk calculation posed in this example pedigree? To do so we need to discuss another aspect of probability, known as Bayes’ theorem.
Bayes’ Theorem. Bayes’ theorem, named for an eighteenth-century mathematician and cleric, offers a formal means to change probability estimates to take into account new information. The process involves changing a “prior probability” based on new data (“conditional probability”). You then calculate a “posterior probability,” a revised estimate.
There are two frequent applications of Bayes’ theorem in clinical genetics: calculating the probability that someone inherited an autosomal dominant disease demonstrating age-dependent penetrance when they are at a given age and remain unaffected (our present example) and calculating the probability that someone is a carrier of a sex-linked or autosomal recessive disease after they have already had some number of unaffected children. You’ll encounter Bayes’ theorem in multiple other contexts throughout medical training. Among other applications, Bayes’ theorem is particularly useful for explaining why screening tests are best used in a population where there is an elevated risk for disease prevalence. Even for the most sensitive and specific screening tests, unless the pre-test probability is elevated, the frequency of false positives will greatly exceed the frequency of true positives. For this course, you will not need to use Bayes’ theorem to calculate exact probabilities; however, we hope that you will gain an intuitive appreciation for its application and understand how it can be used to adjust probabilities in one direction or another.
Returning to the case, it seems reasonable that the longer one who is at risk of inheriting Huntington disease lives without developing symptoms, then the less likely it is that they inherited it. For a 30-year-old person, in the above example, the risk is reduced from 1/2 to 3/7.
SLO 5Explain variable expressivity and what accounts for it
Expressivity – refers to the degree of expression of the phenotype. Unlike penetrance, expressivity takes into consideration varying breadth and/or severity of the clinical features of a disease. The term is most often used in the context of “variable expressivity.” Variable expressivity refers to mutations in the same gene having different clinical consequences, depending upon the person, and can be with reference to severity of one particular manifestation of disease or to the variability of the range of involvement of different tissues and organ systems.
Pleiotropy – Variable expressivity is not to be confused with pleiotropy. Pleiotropy refers to a mutation in a gene that results in multiple phenotypic consequences in diverse tissues.
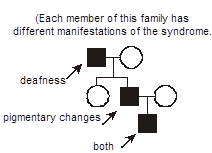 Example of a pleiotropic disorder exhibiting variable expressivity: Waardenburg Syndrome. Each member of this family has different manifestations of Waardenburg syndrome. One has deafness, another has skin and hair pigmentary changes, the third has both deafness and pigmentary changes. Waardenburg syndrome therefore exhibits pleiotropy. Waardenburg syndrome is an autosomal dominant disorder caused by mutations in the gene encoding the PAX3 transcription factor. There is a cellular defect in the migration of neural crest cells during embryogenesis with a resultant phenotype affecting pigmentation and nervous system development. Note that the melanosome pigment cells are derived from the neural crest. Given that neural crest migration contributes to the formation of several organs, it’s easy to understand how mutation of PAX3 has pleiotropic effects. It’s similarly easy to envision one source for variable expressivity of Waardenburg syndrome between different affected individuals. Simply, not everyone will have exactly the same mutation. Individuals from different families will have different mutant alleles (“allelic heterogeneity”). In the particular case of Waardenburg syndrome and many other inherited diseases, however, even individuals within the same family, who would of course be expected to have the exact same mutant allele, demonstrate variable expressivity, with a differing intensity and spectrum of disease.
Example of a pleiotropic disorder exhibiting variable expressivity: Waardenburg Syndrome. Each member of this family has different manifestations of Waardenburg syndrome. One has deafness, another has skin and hair pigmentary changes, the third has both deafness and pigmentary changes. Waardenburg syndrome therefore exhibits pleiotropy. Waardenburg syndrome is an autosomal dominant disorder caused by mutations in the gene encoding the PAX3 transcription factor. There is a cellular defect in the migration of neural crest cells during embryogenesis with a resultant phenotype affecting pigmentation and nervous system development. Note that the melanosome pigment cells are derived from the neural crest. Given that neural crest migration contributes to the formation of several organs, it’s easy to understand how mutation of PAX3 has pleiotropic effects. It’s similarly easy to envision one source for variable expressivity of Waardenburg syndrome between different affected individuals. Simply, not everyone will have exactly the same mutation. Individuals from different families will have different mutant alleles (“allelic heterogeneity”). In the particular case of Waardenburg syndrome and many other inherited diseases, however, even individuals within the same family, who would of course be expected to have the exact same mutant allele, demonstrate variable expressivity, with a differing intensity and spectrum of disease.
Can you think of some explanations? The answers are basically the same as those accounting for incomplete penetrance (but with an additional wrinkle): somewhat weaker effects of so-called “modifying” genes (often called the “genetic background” of an individual) and differing environmental exposures between the individuals. For example, deafness could be influenced by occupational exposure to noise. The additional wrinkle is that there is probably also just an underlying randomness to development, in that neural crest cells migrate somewhat randomly during embryogenesis, so even monozygotic (identical) twins exhibit variable expressivity for Waardenburg syndrome.
Autosomal recessive inheritance. Recessive inheritance implies that both alleles of an autosomal gene must be defective. Autosomal recessive inheritance is often the consequence of a loss-of-function mutation, molecularly resulting from inactivation of the gene. Genes can be inactivated through a variety of different types of mutations. Affected individuals are homozygous for the disease allele and are typically the children of parents who are both unaffected heterozygous “carriers” of the disease allele.
Characteristics of autosomal recessive inheritance
- If it appears in more than one family member, typically it is seen only within one sibship (children of the same parents), not in other generations.
- The recurrence risk for each sib of the proband is 25%.
- More common with consanguinity, especially for rare diseases.
- Usually, males and females are equally likely to be affected.
- New mutation is almost never a consideration.
 Example: cystic fibrosis. Cystic fibrosis (CF) is among the most common autosomal recessive diseases in the European-American and certain other populations. It results from mutations in CFTR, a gene encoding a transmembrane chloride ion channel. The defect in the chloride channel leads to viscous mucous production which, in turn, leads to pleiotropic pathology in primarily three organ systems. Most serious are the pulmonary complications. The bronchioles become progressively dilated, inelastic, and mucous impacted (“bronchiectasis”), and the lungs become recurrently then chronically infected with Pseudomonas, Burkholderia, and other bacterial species. Pancreatic exocrine insufficiency is also frequently seen in CF patients, resulting in gastrointestinal malabsorption. Azoospermia with male infertility is apparent, although, except for the encumbrances of a chronic disease, women with CF can be fertile. There is good “genotype-phenotype” correlation in CF, in that the particular combination of mutant alleles tends to determine both the severity and spectrum of the disease. 70% of the mutant alleles in the European-American population are the “ΔF508” mutation corresponding to a three nucleotide, in-frame deletion that ablates chloride ion channel activity. The mutation deletes a phenylalanine residue (abbreviated as “F” in the single letter code) at amino acid position 508 of the CFTR protein required for trafficking of the channel to the cell surface. The classic ΔF508 homozygote has severe lung disease as well as pancreatic exocrine deficiency.
Example: cystic fibrosis. Cystic fibrosis (CF) is among the most common autosomal recessive diseases in the European-American and certain other populations. It results from mutations in CFTR, a gene encoding a transmembrane chloride ion channel. The defect in the chloride channel leads to viscous mucous production which, in turn, leads to pleiotropic pathology in primarily three organ systems. Most serious are the pulmonary complications. The bronchioles become progressively dilated, inelastic, and mucous impacted (“bronchiectasis”), and the lungs become recurrently then chronically infected with Pseudomonas, Burkholderia, and other bacterial species. Pancreatic exocrine insufficiency is also frequently seen in CF patients, resulting in gastrointestinal malabsorption. Azoospermia with male infertility is apparent, although, except for the encumbrances of a chronic disease, women with CF can be fertile. There is good “genotype-phenotype” correlation in CF, in that the particular combination of mutant alleles tends to determine both the severity and spectrum of the disease. 70% of the mutant alleles in the European-American population are the “ΔF508” mutation corresponding to a three nucleotide, in-frame deletion that ablates chloride ion channel activity. The mutation deletes a phenylalanine residue (abbreviated as “F” in the single letter code) at amino acid position 508 of the CFTR protein required for trafficking of the channel to the cell surface. The classic ΔF508 homozygote has severe lung disease as well as pancreatic exocrine deficiency.
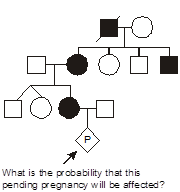
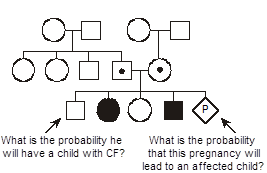
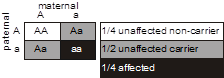 In this pedigree, we wish to know what is the probability that the fetus in the pending pregnancy inherited CF, given that this couple has already had two children affected with CF. Since CF is an autosomal recessive disease, we can infer that both parents are asymptomatic heterozygote carriers of CF mutations. They thus have one normal allele for CFTR and one disease allele. We optionally put a dot in the middle of their pedigree symbols to denote the fact that they are inferred to be obligate heterozygotes. We can set up a Punnett square. In this case we place the carrier mother horizontally on the top and denote her genotype as A/a, where A corresponds to the normal (“wild type”) allele and a represents the mutant allele. We place the father vertically on the left with the same A/a genotype. Since there is a 1/2 probability of segregating either the a or A allele into the sperm or egg during meiosis in the dad or mom, respectively, then we can see that there are four possible outcomes each with equal probability of 1/4. Since there are two different ways to produce a carrier state, the probability that the fetus will be a carrier is 1/4 + 1/4 = 1/2. The probability that the fetus will have genotype a/a and inherit CF is 1/4, and the probability that the fetus will be homozygous for two normal alleles (A/A) is also 1/4. The sum of these mutually exclusive and complete outcomes is one.
In this pedigree, we wish to know what is the probability that the fetus in the pending pregnancy inherited CF, given that this couple has already had two children affected with CF. Since CF is an autosomal recessive disease, we can infer that both parents are asymptomatic heterozygote carriers of CF mutations. They thus have one normal allele for CFTR and one disease allele. We optionally put a dot in the middle of their pedigree symbols to denote the fact that they are inferred to be obligate heterozygotes. We can set up a Punnett square. In this case we place the carrier mother horizontally on the top and denote her genotype as A/a, where A corresponds to the normal (“wild type”) allele and a represents the mutant allele. We place the father vertically on the left with the same A/a genotype. Since there is a 1/2 probability of segregating either the a or A allele into the sperm or egg during meiosis in the dad or mom, respectively, then we can see that there are four possible outcomes each with equal probability of 1/4. Since there are two different ways to produce a carrier state, the probability that the fetus will be a carrier is 1/4 + 1/4 = 1/2. The probability that the fetus will have genotype a/a and inherit CF is 1/4, and the probability that the fetus will be homozygous for two normal alleles (A/A) is also 1/4. The sum of these mutually exclusive and complete outcomes is one.
What is the probability that the unaffected brother will be a carrier? You might be tempted to say 1/2, but that is incorrect. We know that he does not have CF (the black smaller square), so we can eliminate this from the four possible outcomes and cross this off of our Punnett square. This leaves only three possible outcomes in the Punnett square, and we must reset these outcomes so that they sum to one. Since we know that they all originally had equal probabilities, each of the three remaining small squares in our Punnett square should now have a probability of 1/3. Thus, because we know him to be unaffected, the probability that he is a carrier is 2/3. (This is almost certain to be a quiz or board question.)
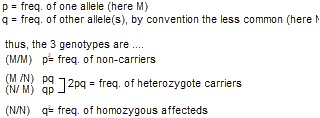 Hardy-Weinberg law. The Hardy-Weinberg law focuses on populations rather than individuals. It is a formalization of the concept that the frequency of alleles in a large population will be constant from one generation to the next, under assumptions that mating is random (the genotype doesn’t influence mate selection) and that the genotype has no selective effect on success at producing offspring. The last three equations below express this law. Again, the sum of the three possible genotypes will equal 1.0.
Hardy-Weinberg law. The Hardy-Weinberg law focuses on populations rather than individuals. It is a formalization of the concept that the frequency of alleles in a large population will be constant from one generation to the next, under assumptions that mating is random (the genotype doesn’t influence mate selection) and that the genotype has no selective effect on success at producing offspring. The last three equations below express this law. Again, the sum of the three possible genotypes will equal 1.0.
A practical implication of the Hardy-Weinberg law is that we can calculate the probability that individuals are carriers of a recessive gene mutation from the prevalence of the disease in the population. Importantly, if these predictions do not match observed population frequencies then we can infer that the mutation does alter genetic fitness (i.e., likelihood of reproduction).
Let’s refer back to the example CF pedigree and ask what is the probability that the brother will have a child with CF. We previously determined that he has a 2/3 chance of being a carrier. We next need to calculate the probability that his mate will be a carrier of CF. We can do this from only knowing the prevalence of the disease in the population, assuming that the mutations do not alter genetic fitness. (Prevalence is the total number of individuals in a population with the condition divided by the total population.) Let’s say that his mate is of broadly European-American ancestry. The prevalence of CF in the European-American population at birth is about 1/2,000. Since everyone with CF must have the homozygous mutant genotype (N/N), we know that q2 = 1/2,000. Thus, q, the frequency of the mutant alleles in the population, equals the square root of 1/2,000, which is about 0.022. We can solve for p explicitly since we know that the sum p + q = 1, but we don’t really need to do this for these kinds of calculations.
It turns out that since q is usually so small, meaning p is usually so close to one, we can just approximate p ≈ 1 when using the Hardy-Weinberg equation.
The frequency of heterozygote carriers in the population is thus just 2pq, which is approximately equal to 2q, which is 0.044. So about 4.4% of the European-American population are CF carriers, corresponding to about 1/23 European-American people. (Note that even for a somewhat rare autosomal recessive 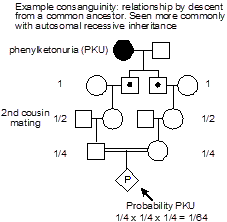 disease like CF, heterozygote carriers are fairly common, because 2pq >> q2.) Thus, to calculate the probability that the brother in the above pedigree will have a child with CF, it is his chance of being a carrier (2/3) times the chance that, if he were a carrier, that he would transmit the mutant allele during the meiosis producing his sperm (1/2) times the chance that his mate, randomly selected from the European-American population, would also be a carrier (1/23) times the chance that—should she be a carrier—that she would transmit the mutant allele during the meiosis producing her egg (1/2). And the answer is 2/3 × 1/2 × 1/23 × 1/2 = 0.008 or 0.8%. CF generally adheres to Hardy-Weinberg equilibrium. Negative effects on genetic fitness are generally restricted to the small q2 population of affected homozygotes with the mutation, and, if anything, the much larger 2pq population of unaffected heterozygote carriers may actually have a reproductive advantage, for various postulated reasons.
disease like CF, heterozygote carriers are fairly common, because 2pq >> q2.) Thus, to calculate the probability that the brother in the above pedigree will have a child with CF, it is his chance of being a carrier (2/3) times the chance that, if he were a carrier, that he would transmit the mutant allele during the meiosis producing his sperm (1/2) times the chance that his mate, randomly selected from the European-American population, would also be a carrier (1/23) times the chance that—should she be a carrier—that she would transmit the mutant allele during the meiosis producing her egg (1/2). And the answer is 2/3 × 1/2 × 1/23 × 1/2 = 0.008 or 0.8%. CF generally adheres to Hardy-Weinberg equilibrium. Negative effects on genetic fitness are generally restricted to the small q2 population of affected homozygotes with the mutation, and, if anything, the much larger 2pq population of unaffected heterozygote carriers may actually have a reproductive advantage, for various postulated reasons.
Example: hemochromatosis. It should be emphasized that incomplete penetrance and variable expressivity, as well as pleiotropy, are all seen with autosomal recessive disease (or sex-linked recessive inheritance, which we will discuss shortly), just as is the case with autosomal dominant inheritance. A good example of an autosomal recessive disease that demonstrates both incomplete penetrance and variable expressivity (in addition to pleiotropy) is hemochromatosis. Hemochromatosis, or “iron overload syndrome” is among the most common recessive diseases in individuals of European-American descent. It results from mutations in a gene, HFE, involved in iron transport via binding to the transferrin receptor. A single allele (C282Y), resulting in a cysteine-to-tyrosine missense amino acid substitution at the 282nd position of the polypeptide, accounts for about 88% of all disease-associated alleles in this population. (A milder allele, H63D, accounts for most of the rest of the mutations in this population.) The disease clinically manifests with cirrhosis and consequent risk for hepatocellular carcinoma, characteristic arthritis, bronzing of the skin, cardiomyopathy, diabetes mellitus, and, in males, testicular atrophy. Elevation of the serum iron carrier protein, ferritin, serves as a laboratory marker of disease. Hemochromatosis is simply treated, if diagnosed early enough, with phlebotomy (blood draw), since red blood cells represent the major form of storage of iron in the body. It used to be definitively diagnosed through liver biopsy and corroborating laboratory studies, including increased levels of serum ferritin and transferrin, but DNA diagnostic studies are now the gold standard.
Not everyone who is homozygous for the most common mutant allele will develop symptoms of hemochromatosis. Thus, this disease demonstrates incomplete penetrance. The reasons for this are that not everyone is exposed to the same environmental and genetic factors. The environmental factors would include dietary iron intake. Other genetic factors regulating iron metabolism modify hemochromatosis risk. Furthermore, this disease demonstrates age-dependent penetrance, merely because it takes some time for the toxic accumulation of iron in organs. The disease also demonstrates sex-dependent penetrance. Since females are more likely to menstruate and have an average lower red blood cell mass, their total body iron stores tend to be less than those of non-menstruating males, and they are less likely to develop organ toxicity. Furthermore, the disease demonstrates variable expressivity, in that not everyone who has hemochromatosis will exhibit similar clinical severity or patterns of organ system involvement. For example, obese individuals and those with other hereditary risk for diabetes mellitus might be more likely to develop this complication of the disease. Individuals who consume ethanol may be more likely to sustain liver damage. Finally, individuals who are “compound heterozygotes” for H63D/C282Y alleles (as opposed to C282Y homozygotes), typically have milder disease, while H63D homozygotes are often unaffected.
Consanguinity. “Consanguinity” refers to relationship by descent from a common ancestor (also known as “inbreeding”). Consanguinity is a concern in autosomal recessive disease, because if this is a rare disease (due to an infrequent allele), then the disease will occur more commonly in individuals whose 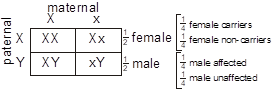 parents are related. Consanguinity is noted on a pedigree by two horizontal lines between the male and female partner.
parents are related. Consanguinity is noted on a pedigree by two horizontal lines between the male and female partner.
Always consider the possibility of consanguinity in the approach to care of a patient with autosomal recessive diseases, especially when the disease is rare.
Example: phenylketonuria. In this example, we can see how there is an increased probability of a recessive disease, here phenylketonuria (PKU), occurring in a pregnancy resulting from a consanguineous mating. PKU most commonly results from deficiency of phenylalanine hydroxylase, which is involved in the metabolism of the amino acid phenylalanine. This leads to the accumulation of neurotoxic levels of phenylalanine. Clinical manifestations include intellectual disability, seizure disorder, a “mousy” odor, and hypopigmentation of skin and hair. Children who inherit the metabolic defect, but who avoid dietary exposure to high concentrations of phenylalanine, however, are essentially normal. PKU screening at birth has therefore become routine. It is important to ask about the possibility of consanguinity when taking a family history, especially when considering the possibility of an autosomal recessive disease. For the case of PKU, its occurrence is relatively uniform across the globe; however, an increased frequency has been seen in individuals from the Romani population. The diagnosis of a rare disease in an ethnically isolated or otherwise consanguineous pedigree should make one think about the possibility of autosomal recessive inheritance.
Sex-linked recessive. Sex-linked recessive inheritance occurs when the mutated gene resides on the X chromosome and acts recessively. This is also known as “X-linked recessive inheritance.” Since females have two copies of the X chromosome, in order to be affected, both copies of the responsible gene must correspond to a disease-causing allele. This is much less likely than in the situation for males, in which there is only a single X chromosome. Males are therefore considered “hemizygous” for the X chromosome. Males inherit their single X chromosome from their mother, so sex-linked recessive disease follows a maternally inherited distribution pattern in the family.
Characteristics of sex-linked recessive inheritance
- Males are more commonly affected than females.
- The gene responsible is transmitted from an affected male through his female offspring, who are seldomly affected. Each female offspring is an obligatory heterozygous carrier. Each of the carrier female offspring’s male children has a 50% chance of inheriting it.
- No male-to-male transmission occurs.
- The affected males in a pedigree are usually related through females.
- Heterozygous female carriers are usually unaffected, but some may express the condition with variable severity (called skewed “Lyonization” or “skewed X-chromosome inactivation”).
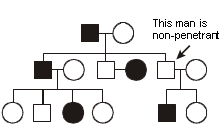
Example: hemophilia A. Hemophilia A is among the most common of sex-linked recessive diseases. It results from a deficiency of factor VIII, a component of the blood clotting cascade. Most of the bleeding is into joint spaces or into the gut. Males who have a factor VIII mutation are affected because they are hemizygous for the gene, having only one copy of the X chromosome where the gene resides. Females who have a factor VIII mutation are usually unaffected. Although they may have reduced levels of factor VIII activity (resulting from the phenomenon of X chromosome inactivation, described below), this is usually sufficient to prevent development of clinical symptoms.
Here is the pedigree of an affected family. What is the probability that the fetus in the pending pregnancy will have inherited hemophilia A? We can assume that the pregnant mother is a carrier. She has already had one affected male child, and there is an extensive history of hemophilia A in males in the family. (As you’ll see later, if there were only this one affected male child, we could not 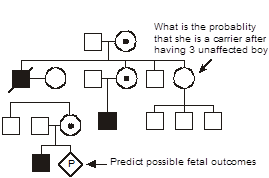 exclude the possibility that he is the result of a new mutation.) Since the mother is a carrier, then we can deduce that she has one normal factor VIII allele and one mutant factor VIII allele. The probability that she will transmit the mutant allele during oogenesis is therefore 1/2. The probability that the fetus will inherit the mutant factor VIII allele is therefore also 1/2. In this case, the sex of the fetus is important. If the fetus inherits an X chromosome from the father and is female, then she will probably not be affected with hemophilia A (although there are some exceptions that we will come to). So, we need only be concerned with the possibility that the fetus inherits a Y chromosome from the father and is a male—an event with probability of 1/2. The product of these two independent events, the 1/2 probability that the mother will contribute her mutant factor VIII allele to the egg and the 1/2 probability that the father will contribute a Y and make the conceptus a male, is 1/4, and this is the probability that the fetus will be a male inheriting hemophilia A. Again, one can draw out a Punnett square, but we should start to be able to see these things by now without the aid of such didactic tools.
exclude the possibility that he is the result of a new mutation.) Since the mother is a carrier, then we can deduce that she has one normal factor VIII allele and one mutant factor VIII allele. The probability that she will transmit the mutant allele during oogenesis is therefore 1/2. The probability that the fetus will inherit the mutant factor VIII allele is therefore also 1/2. In this case, the sex of the fetus is important. If the fetus inherits an X chromosome from the father and is female, then she will probably not be affected with hemophilia A (although there are some exceptions that we will come to). So, we need only be concerned with the possibility that the fetus inherits a Y chromosome from the father and is a male—an event with probability of 1/2. The product of these two independent events, the 1/2 probability that the mother will contribute her mutant factor VIII allele to the egg and the 1/2 probability that the father will contribute a Y and make the conceptus a male, is 1/4, and this is the probability that the fetus will be a male inheriting hemophilia A. Again, one can draw out a Punnett square, but we should start to be able to see these things by now without the aid of such didactic tools.
Now let’s ask a more difficult question. What is the probability that the woman indicated by an arrow in the pedigree is actually a carrier of hemophilia A after giving birth to three unaffected children? At birth, she was at 1/2 risk of having inherited the mutant allele from her carrier mother. However, as she keeps giving birth to unaffected males, we might begin to doubt that she is a carrier. The more unaffected males she produces, then the less likely it is that she actually inherited carrier status for hemophilia A. It is almost as if she is revealing to us her hand of cards by letting us know what remains in the deck, card by card. To precisely calculate her risk requires use of Bayes’ theorem. We won’t do so here because you won’t need to perform these calculations for the course. However, in case you are interested, the risk that she is a carrier after having given birth to three unaffected males is reduced to 1/9.
To try to get an intuitive grasp for this question, think of the extreme case. What if she were initially at 1/2 risk to be a carrier, but she had 25 unaffected male children? Of course, at this point we would begin to seriously doubt that she inherited the mutant factor VIII allele.
X chromosome inactivation. In XX female individuals, one of the two copies of the X chromosome is largely inactivated, in a process known as “Lyonization.”
Lyonization – a term used for the random inactivation of one of the X chromosome in each cell of a female is named after Mary Lyon, who discovered X inactivation. The word is most often used when there is a “skewed” or “unfavorable” pattern of X chromosome inactivation, such that the female is at least partly affected for a sex-linked recessive disorder.
 Females possess two copies of X chromosome genes since they have two X chromosomes. In contrast, males have only one copy of genes residing on the X chromosome since they only have one X chromosome. The Y chromosome, for the most part, does not contain any of the same genes. The evolutionary rationale for inactivating one of the X chromosomes in females is presumably as a mechanism of “dosage compensation” to ensure that both sexes have the same number of functioning alleles for genes on the X chromosome. The particular X chromosome that is transcriptionally silenced in any given cell well be condensed, making it visible even during “interphase” of the mitotic cell cycle, when chromosomes are ordinarily elongated and not microscopically visible. The name for the condensed microscopic appearance of the X chromosome is the “Barr body.” Females ordinarily have one Barr body per cell, whereas males have zero. Since X chromosome inactivation in humans is randomly distributed in early embryonic development throughout tissues, skewing of inactivation might result in a segmental pattern of distribution of expression of the mutant gene and resulting clinical phenotype as shown in the middle and right figures. Some women could have some locally uneven patterns of X inactivation, merely due to random statistical variation. There are a few genes in females, however, that do escape X inactivation, even though the rest of the chromosome is shutdown.
Females possess two copies of X chromosome genes since they have two X chromosomes. In contrast, males have only one copy of genes residing on the X chromosome since they only have one X chromosome. The Y chromosome, for the most part, does not contain any of the same genes. The evolutionary rationale for inactivating one of the X chromosomes in females is presumably as a mechanism of “dosage compensation” to ensure that both sexes have the same number of functioning alleles for genes on the X chromosome. The particular X chromosome that is transcriptionally silenced in any given cell well be condensed, making it visible even during “interphase” of the mitotic cell cycle, when chromosomes are ordinarily elongated and not microscopically visible. The name for the condensed microscopic appearance of the X chromosome is the “Barr body.” Females ordinarily have one Barr body per cell, whereas males have zero. Since X chromosome inactivation in humans is randomly distributed in early embryonic development throughout tissues, skewing of inactivation might result in a segmental pattern of distribution of expression of the mutant gene and resulting clinical phenotype as shown in the middle and right figures. Some women could have some locally uneven patterns of X inactivation, merely due to random statistical variation. There are a few genes in females, however, that do escape X inactivation, even though the rest of the chromosome is shutdown.
At a molecular level, the inactive X chromosome expresses a unique gene, XIST, encoding a non-translated RNA. The XIST RNA physically accumulates along the inactive X chromosome causing inactivation of most of the genes on this chromosome and leading to its condensation into a Barr body. Somewhat paradoxically, the XIST gene is not expressed from the active X chromosome (whereas the other genes on this copy of the X chromosome are expressed). On the active X chromosome, the promoter of the XIST gene is heavily methylated at CpG sequences, thereby silencing its expression.
Consider factor VIII in hemophilia A. Most of the factor VIII production occurs in the liver where it is secreted into the peripheral blood stream. Extreme skewing in X inactivation is uncommon for hemophilia A. This is because it does not really matter whether there is a locally uneven pattern of X inactivation in the liver cells. On balance, close to half of the hepatocytes will be producing factor VIII, and it will all get averaged out upon secretion into the blood. A completely different situation pertains to a protein that is not secreted, but rather whose effect is local to the cell that produces it. A good example is Duchenne muscular dystrophy, a sex-linked autosomal recessive severe form of muscular dystrophy primarily affecting males but for which females are sometimes symptomatic. In the case of Duchenne muscular dystrophy, the defect is in a protein, dystrophin, which connects the cytoskeleton of a muscle fiber to the surrounding extracellular matrix through the cell membrane. If there were a skewed pattern of X inactivation in a particular muscle, then we might expect that muscle to be weak and there to be a focal, segmental pattern of weakness.