
2 Genetics Introduction
Session Learning Objectives and a quick synopsis (see main text for explanations):
The goal of this chapter is to provide a brief overview of genetic concepts.
SLO 1 Explain the different ways genetics contributes to disease risk
Distinguish single-gene Mendelian inheritance (autosomal dominant, autosomal recessive, or sex-linked recessive) from complex polygenic inheritance (which contributes to most common diseases). Differentiate germline versus somatic mutation.
SLO 2 Record and interpret a human pedigree using standard symbols
Learn to draw and read the genetic shorthand for a family tree with squares (males) and circles (females), filled (affected) and open, and so forth, to trace the inheritance of genotypes and phenotypes.
SLO 3 Define the relationship between a gene and its phenotype
Define gene, locus, genotype, phenotype, allele, and distinguish homozygosity from heterozygosity.
SLO 4 Apply probabilities to independent vs. dependent genetic events
For simple genetic outcomes, learn when to multiply (independent events) and when to add (mutually exclusive events) probabilities.
Main text
The human genome is comprised of DNA, organized into genes, and carried on 46 chromosomes. Molecular biology encompasses how the DNA is transcribed into RNA and how RNA is translated into proteins. Human genetics is the study of how our genome and its variants give rise to our human form and its variation and how the genomes and variations of one generation are recombined and passed on to the next generation—inheritance.
SLO 1 Explain the different ways genetics contributes to disease risk
Our genes and chromosomes are constantly subject to change. Genetic variations that are due to changes in the “germline” genome will be passed from one generation to the next. They are called hereditary variations. They contrast with genetic variations acquired post-zygotically in any of the vast number of cells not contributing to gametes. Such mutations are called somatic mutations. They cannot be passed between generations. Most diseases are not primarily hereditary. Nevertheless, genetics is hugely important in human disease, and for almost any disorder, inherited risk factors, acting in isolation or in concert with the environment, can be identified.
Normal genetic variation versus disease. Most genetic variation is responsible for differences in traits distinguishing one person from the next but can also influence vulnerability to disease. Moreover, genetic factors likely have played different biological roles in different places and times throughout history as our environment has changed. Genetic variation that at one time may have conferred reproductive advantages may be harmful for modern life.
Single-gene Mendelian disorders. “Single gene” or “Mendelian” disorders are ones in which mutations in just one gene can be passed down with a predictable pattern of disease from one generation to the next. Such disorders often have dramatic clinical consequences. About 2-5% of the population has a Mendelian disorder. Many, such as cystic fibrosis, can be apparent at birth whereas others, such as Huntington Disease, are more likely to appear with aging. We will be distinguishing between single-gene Mendelian disorders and complex disorders, arising from common genetic variation of smaller effect size distributed throughout the genome and working in concert with the environment.
To understand inheritance, we need to remember that each gene lies on a specific chromosome, either on a sex chromosome or on one of the 22 pairs of autosomes (non-sex chromosomes). Normally, there are two similar copies (alleles) of each autosomal gene. For an autosomal dominant disorder, only one of the two copies need sustain a pathogenic mutation in order to cause disease, whereas, for an autosomal recessive disorder, mutations in both copies are required to cause disease. During the formation of a gamete, one of the two alleles is randomly included in each egg or sperm. Since the distribution is random, we have to think about probability theory to calculate the risk of transmitting a disorder. The risk of transmitting an autosomal dominant disorder from affected parent to child is 1/2 and is independent of the sex of the parent. The risk of transmitting an autosomal recessive disorder when each parent is a carrier is 1/4. Sex-linked recessive disorders arise from mutations on the X chromosome. Females have two X chromosomes and are seldomly affected. However, for female carriers, the risk of conceiving an affected male (XY chromosome composition) or a carrier female (XX chromosome composition) is 1/2. (This is just a brief introduction. We will be talking about each of these forms of Mendelian inheritance in greater detail later.)
In general, for Mendelian disorders, there is a diversity of mutations within the causative gene across the affected population. The mutation responsible for the disorder in one family may not be the same as one causing disease in a different family.
Exceptions are ancestral “founder” mutations that may be over-represented among certain populations. The autosomal recessive beta-chain hemoglobin mutation that causes sickle cell anemia is one example. In the carrier state, it confers relative resistance to malaria that bestows a selective advantage throughout regions of the world where malaria is prevalent. Another example involves three Ashkenazi Jewish founder mutations in the BRCA1 and BRCA2 DNA repair factors, which are frequent in contemporary populations simply because in an early population bottleneck for this community there were only a few founding ancestors. Apparently, by chance they happened to possess these three mutations.
Overall, disease alleles for Mendelian disorders are rare although they may be more prevalent for certain diseases in certain populations. This is in contrast to complex disorders, where the disease-associated alleles are common, albeit of much weaker effect.
By and large for Mendelian disorders, most pathogenic mutations alter the amino acid sequence of the polypeptide encoded by the gene or lead to its loss of production. For complex disorders, mutations often have less dramatic effects on gene function, are better tolerated and therefore prevalent across the population, and consequently exert their effects more subtly, often by influencing gene regulatory regions.
Complex disease. In contrast to Mendelian disorders, most common diseases are the result of additive effects of common genetic variants conferring variable degrees of risk interacting with lifestyle and environment—so called “complex” disorders. (“Multifactorial” and “polygenic” are synonyms for “complex.”) Examples include hypertension, diabetes mellitus, atherosclerotic vascular disease, autoimmune disorders, etc. For any disease, there are typically dozens of variant alleles for any of multiple associated genes, some of which increase risk and some of which decrease risk. Each variant may interact with environmental factors, such as diet and tobacco smoking, in different ways. Common diseases often cluster in families because these variants, along with environmental factors and the behaviors associated with them, are shared between relatives, although in less predictable ways than the clear-cut patterns apparent for Mendelian disorders. Some disorders, for example hypercholesterolemia, can result from either rare single-gene disorders inherited from just one parent or, more commonly, due to inheritance as a complex trait involving multiple genetic variants inherited from both parents and influenced by non-genetic factors, such as diet.
Chromosomal and other genomic disorders. Chromosomes can undergo physical alterations. These changes can be microscopically visible or molecularly detectable duplications, deletions, insertions, inversions, translocations of segments between chromosomes, or other sorts of structural rearrangements that can disrupt the protein-coding and/or regulation of multiple genes at once. Copy number variation is a type of structural variation in which a stretch of DNA is duplicated or deleted. Autosomal chromosomal abnormalities and copy number variants can produce dramatic physical phenotypes. Abnormalities of sex chromosomes typically have less severe consequences. About half of all first-trimester spontaneous abortions (miscarriages) contain chromosomal abnormalities.
Somatic genetic variation. Mutations and chromosomal abnormalities can occur in the “germline” and will typically be present in all the cells of the body. Or they can be “acquired” (“somatic”) such that they arise post-zygotically and are restricted to a subpopulation of cells. Somatic genetic variation is responsible for cancer but contributes to other disorders, as well as aging. Some inherited cancer predisposition syndromes depend upon an inherited gene for cancer risk, with the actual onset and tissue distribution of the resulting cancer determined by the occurrence of somatic mutations in the same or other genes.
Genetic testing depends on the type of genetic variation. Mendelian disorders exhibit predictable patterns of inheritance and can often be directly tested for by gene sequencing tests. Evaluating the genetic underpinnings of common disease is more challenging and requires genome-wide based approaches. Although not typically used in a clinical setting, such testing has become popular among consumers, where considerable confusion by patients and providers surrounds their use. Chromosomal and copy number variant disorders use a unique collection of technologies for their diagnosis. The identification and the role of acquired mutations in diagnosis and treatment of cancer continues to evolve and is increasingly relied on to personalize molecular therapies. Historical routes to the discovery of the molecular genetic basis of these different sorts of disorders has varied. Genetics is one of the most rapidly changing fields, contributes to the practice of multiple areas of medicine, and makes increasingly greater contributions to improving and individualizing the care of patients.
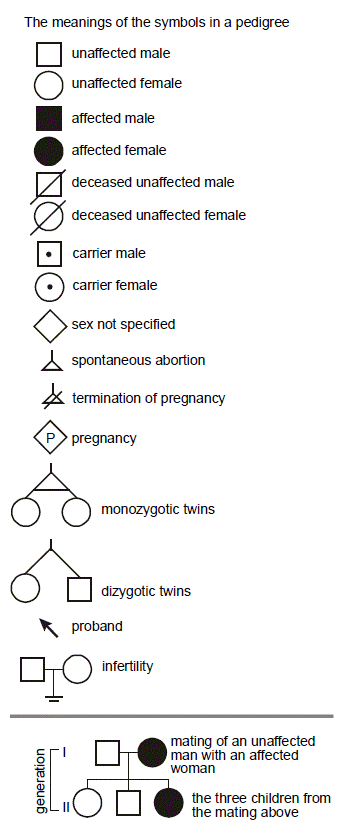
SLO 2 Record and interpret a human pedigree using standard symbols
To simplify recording family history, we make use of symbols. Males are squares. Females are circles. Death is noted with a slash. Horizontal lines connect reproductive partners. Vertical lines indicate parent-child relationships. Diamonds are used when sex is unspecified. Putting a number in the middle of the symbol can denote multiple individuals. The notation for a fetus or pending pregnancy is a diamond with a “P” in the middle. Spontaneous abortion and termination of pregnancy are often shown with a small triangle, without and with a slash, respectively. The “proband” (usually the patient) is the individual who brought the family to attention and is highlighted with an arrow. Dizygotic (fraternal) twins have a forked line connecting them, while monozygotic (identical) twins are joined with an additional horizontal line. The symbols are shaded to indicate a disease. When more than one disease is present, the symbols may be sectored like pieces of pie. We will employ these symbols in this course. For a typical family history, recorded in a medical record, pedigrees of this sort had been considered overkill; however, readily available modules for the electronic health record can automatically construct a detailed pedigree from a short questionnaire completed by patients.
SLO 3 Define the relationship between a gene and its phenotype
IMPORTANT DEFINITIONS:
Gene – The basic hereditary unit, initially defined by phenotype. By molecular definition, a DNA sequence required for production of a functional product, usually a protein, but rarely an untranslated RNA.
Locus – Literally a “place” on a chromosome or DNA molecule, used fairly interchangeably with “gene” and sometimes used to refer to a collection of closely spaced genes.
Genotype – An individual’s genetic constitution, either collectively at all loci or more typically at a single locus or even at a particular position within a gene.
Phenotype – Observable expression of genotype as a trait or disease.
Allele – One of the alternate versions of a gene present in a population. An over-simplification is that there are just two, “normal” (wild type) and mutant. Of course, for any gene there are typically multiple different wild type alleles, distinguishable by clinically inconsequential “polymorphisms,” and many different potential mutant alleles.
Homozygosity vs. Heterozygosity – For all autosomal (non sex chromosome) genes, there are two alleles, one inherited from the mother and one inherited from the father. If both versions are the same (both wild type or both mutant), we use the term “homozygous,” whereas if each allele is different (one wild type and one mutant) we use the term “heterozygous.”
 Genes were originally defined by Gregor Mendel, a nineteenth century Austrian monk, who formulated the basic rules of inheritance while observing peas, as the theoretical unit responsible for traits (“phenotype”). He correctly deduced that each parent contributes one copy of each of the offspring’s two copies of a gene for most traits and that there are different versions of a gene, known as “alleles,” within a population. Some of these alleles function “dominantly,” in that they are sufficient to cause the phenotype regardless of the allele contributed by the other parent. Other alleles act “recessively” and require that the opposite parent contribute a similarly abnormal allele for the phenotype to be manifest.
Genes were originally defined by Gregor Mendel, a nineteenth century Austrian monk, who formulated the basic rules of inheritance while observing peas, as the theoretical unit responsible for traits (“phenotype”). He correctly deduced that each parent contributes one copy of each of the offspring’s two copies of a gene for most traits and that there are different versions of a gene, known as “alleles,” within a population. Some of these alleles function “dominantly,” in that they are sufficient to cause the phenotype regardless of the allele contributed by the other parent. Other alleles act “recessively” and require that the opposite parent contribute a similarly abnormal allele for the phenotype to be manifest.
SLO 4 Apply probabilities to independent vs. dependent genetic events
Mendelian inheritance involves three simple statistical rules:
- Multiply probabilities for independent events.
- Add probabilities for mutually exclusive events.
- The total probability for all possible outcomes must sum to one.
One example is a coin toss. The probability of heads or tails is one-half and is independent of any prior tosses. “Chance has no memory.” Even if there had been a run of heads, the next toss is no more or less likely to be a tails than if there had been a run of tails—unless something is wrong with the coin! The likelihood of having two consecutive heads is 1/2 × 1/2 = (1/2)2 = 1/4; the probability of having three tails in a row is just 1/2 × 1/2 × 1/2 = (1/2)3 = 1/8. When you toss a coin, the two possible outcomes are heads or tails; therefore, the sum of the probabilities of each outcome must add up to a total probability of one. So, for a coin tossed three times consecutively, each of the eight possible outcomes occurs with an equal probability of 1/8.
What’s the chance that a pregnant XX female will bear an XY male conception? That’s an easy one; it’s about 1/2. Same question but for an XX female conception? Same answer (1/2). Since these are mutually exclusive probabilities, we know that the probability of conceiving either an XY male or an XX female must be one. Reassuringly, that also happens to be the sum of 1/2 plus 1/2. (For the sake of this exercise to illustrate simple probability concepts we consider only binary, chromosomally defined sex and exclude other chromosomal and non-chromosomal sex variation.)
What’s the probability that a pregnant XX female will conceive two consecutive XY males? Since these are independent events, we know that it must be 1/2 times 1/2, which equals 1/4. What about two XX females in a row? Same thing. How about one XY male and one XX female? For two children you can only have either two XY males, two XX females, or one of each, so the probability of an XY male and an XX female must equal the probability of not having two males or two females (since these are mutually exclusive events summing to one), and that is merely 1 – 1/4 – 1/4 = 1/2. But why is it twice as likely to have an XY male and an XX female as having two XY males or two XX females? That’s because there’s two different sequences in which to have an XY male and an XX female. The XY male could come first, followed by an XX female, and the chance of that sequence happening is also 1/2 × 1/2 = 1/4. Or, the XX female could arrive first, followed by an XY male, and the chance of that sequence happening is the same.
Feedback: