
5 Complex Traits, Meiosis, Genome Wide Association Studies (GWAS)
Session Learning Objectives and a quick synopsis:
Nearly all diseases have a genetic component, though most are not highly penetrant, single gene, Mendelian disorders. Instead, common diseases are the result of many common genetic variants with small effect size that are spread throughout the genome. Some of these variants increase risk while others lower risk. The sum of these inherited factors, in combination with lifestyle/environment (which can also be shared among families and populations) influence susceptibility to most diseases.
SLO 1 Define what is meant by “complex genetics”
Most common diseases are inherited through “complex genetics,” and are not caused by just one highly penetrant gene mutation. Instead, they result from a combination of common population variants in multiple genes, acting in concert with environmental factors.
SLO 2 Describe normal genetic variation in the human population
When comparing the genomes of any two people, most of the differences are due to single nucleotide polymorphisms (SNPs). On average, there is a SNP differentiating any two unrelated people about every thousand base pairs or so. Any two unrelated people differ by about 5-10 million SNPs. Most of the SNPs and other types of sequence variants in the genome that differentiate people are of ancient human origin and occur commonly within and across populations.
SLO 3 Explain how natural selection influences disease gene frequencies
Human populations are subject to evolutionary pressures. Deleterious mutations tend to not be maintained due to their harmful effects, whereas beneficial mutations are selected for. However, the same mutation can be beneficial or harmful, depending upon gene dosage (heterozygous vs. homozygous) and environmental context. A classic example is sickle cell disease, an autosomal recessive disease that nevertheless confers malarial resistance to asymptomatic carriers and is therefore maintained in the population.
SLO 4 Explain how meiosis increases genetic diversity
Recombination and independent assortment during meiosis produces new gene combinations in offspring, thereby adding to population diversity with each new generation.
SLO 5 Explain the concept of linkage disequilibrium and haplotypes
Genetic variants close by one another on the same chromosome tend to be inherited together, because the probability of a recombination event occurring between them during meiosis is low—a phenomenon known as “linkage disequilibrium.” Resulting genetic variants commonly inherited across a chromosomal region shared between people within a family or at the population level define a “haplotype.”
SLO 6 Describe the origins of the haplotype block structure of the genome
Over time, meiotic recombination whittles down the length of each haplotype and reduces ancestral contributions to small chromosomal “blocks” of a few thousand contiguous base pairs. Because these chromosomal regions cannot further recombine, they reach a lower size limit in which all the genetic variants contained within the region are inherited together. Our genomes therefore represent a mosaic derived from ancient ancestors, in which the contributions from any ancestor from more than just a few generations ago is indistinguishable.
GWAS is an experimental tool used to identify common variants with small effect sizes, collectively contributing to disease risk. It involves a simple association test to see if particular SNPs representative of certain ancestral haplotype blocks are found more commonly in people with a certain trait compared to controls.
Main Text:
SLO 1 Define what is meant by “complex genetics”
In contrast to Mendelian disorders such as sickle cell anemia, Marfan syndrome, or cystic fibrosis, in most cases common diseases such as hypertension, atherosclerotic vascular disease, diabetes mellitus, and mental illness cannot be explained by mutations in a single gene. Instead, they are usually associated with the effects of multiple genes interacting in combination with lifestyle and environmental factors. (However, for almost any common disease, such as hypercholesterolemia or cancer, there are the somewhat rare families who are an exception and inherit the disorder due to a causative mutation in a single gene.) This is what is meant by “complex genetics” or “multifactorial” or “polygenic” disorders.
Heritability. For a complex trait, its “heritability,” which is the fraction of phenotype variability attributable to underlying genetic variation, can be measured through several different epidemiological approaches. One such approach is to study twins.
Twin studies. About 1/100 births are twins. Dizygotic (DZ), or fraternal twins, result from the fertilization of two different eggs, whereas monozygotic (MZ), or identical twins, are the product of a division of the developing embryo. Dizygotic twins share 50% of their DNA (just as do ordinary siblings) whereas monozygotic twins share 100%. Geneticists use the concept of concordance in twin studies, where if both twins share the same trait or both are unaffected they are said to be concordant. Measuring the difference in concordance frequency for a trait between monozygotic and dizygotic twins can separate genetic from environmental factors (under the assumption that the environments are the same for both monozygotic and dizygotic twins).
Heritability of some common traits. For example, the concordance frequency for bipolar affective disorder for monozygotic twins is 79%. Hence, in a pair of monozygotic twins, if one twin is affected then 79% of the time the other twin will be, too. In contrast, the concordance frequency for bipolar affective disorder is only 24% for dizygotic twins. For a highly penetrant autosomal dominant disorder unaffected by environment, concordance for monozygotic twins would be 100%, and concordance for dizygotic twins would be 50%. Thus, it can be concluded that, at the population level, there is a strong genetic contribution to the risk for bipolar affective disorder, but it stops short of what would be expected for a single-gene disorder, and there is also evidence for contributions from environmental factors. In contrast, the concordance frequency for measles, an infectious disease caused by a single-stranded RNA virus, for which there could nevertheless be some degree of genetic risk for vulnerability, is 95% in monozygotic twins and 87% in dizygotic twin pairs. This supports the fact that it is largely the shared environment between twins, rather than the extra 50% identity in genetics, that is mainly responsible for measles infections.
Threshold concept for complex inheritance. The threshold model for complex genetics posits that there is a certain cumulative “red line” of environmental and genetic risk factors that must be crossed to develop a particular disease.
For a complex disease, a threshold level of environmental and genetic risk factors must be crossed to develop the disease. Not all genes contribute equally to risk in all people for a complex disease.
Sex–dependent thresholds. The threshold for developing the disease may differ between males and females. For example, coronary artery disease is a complex disease resulting in rare circumstances from the influence of a single gene (such as mutations in the LDL receptor in familial hypercholesterolemia), but more often from a polygenic contribution (i.e., multiple genes influencing cholesterol, triglycerides, homocysteine, etc.) as well as environmental factors (such as diet, tobacco smoking, and exercise). For any given level of genetic and environmental risk, however, males have more coronary artery disease, at least in part because of the protective effects of estrogen in females.
There is an interesting consequence of the fact that the risk threshold differs between the sexes: the recurrence risk in children will be greater for the children of the less susceptible sex.
For example, if one’s mother has coronary artery disease, that person is at greater risk for developing coronary artery disease (regardless of their sex), than if their father had coronary artery disease. Since females have a higher threshold for developing coronary artery disease, they must therefore have more risk factors (genetic and environmental), overall, and are consequently likely to specifically have more genetic risk factors that one is capable of inheriting. A person is therefore more likely to inherit more genetic risk factors for coronary artery disease from one’s mother than they would if it were their father who were affected with coronary artery disease.
What is needed to understand the basis of complex genetic disorders. Before we can understand the molecular genetic basis of complex traits, we need to dive a little more deeply into some fundamental aspects of human genetics, as it relates to the sources of human genetic variation and how genetic variation arising in any one individual may be represented across a population in subsequent generations over time.
SLO 2 Describe normal genetic variation in the human population
The sequence of our genome contributes to what makes us unique—and what we have in common with other human beings.
The human “diploid” genome is about 6.6 Gb (billion base pairs, “gigabases”). Recall that there are 23 pairs of human chromosomes, one member of each pair being inherited from each parent. Consequently, the “haploid” genome, defined as the genome in a gamete (egg or sperm), is 3.3 GB. For most genes there is one maternal and one paternal copy except for the small circular mitochondrial genome (16,569 base pairs) and a few genes unique to the X chromosome and far fewer genes unique to the Y chromosome.
The most common type of genetic difference between any two people involves single base-pair differences, otherwise known as a “single nucleotide polymorphism” (or, more commonly, “SNP”).
Mutation – A change in the DNA sequence usually conferring a deleterious effect (at least in a medical context). It may be present in the germline causing an inherited disorder or it may be acquired post-zygotically in somatic tissues, for example, in cancer.
Polymorphism – A DNA sequence difference usually of no pathological consequence. The difference between a mutation vs. polymorphism gets into rhetorically muddy waters when it comes to sequence differences contributing to traits, such as blue eyes vs. brown, with little meaningful medical significance. In these circumstances, the use of the term “variant” may be preferred.
Variant of undetermined (or unknown) significance (VUS) – This is a term nearly always used in the context of genetic testing. Genetic testing often turns up a VUS. Such a DNA sequence difference may be unique to the tested individual, or at least not commonly found in other people, and its properties may not be clearly pathogenic and/or not previously shown to segregate with disease. Finding a VUS on genetic testing is another way of saying, “we found something but don’t have sufficient information to determine its clinical implications, if any.”
Any two unrelated people differ by about 5-10 million SNPs. On average, there is a SNP differentiating any two unrelated people about every thousand base pairs or so.
The vast majority of SNPs and other types of sequence variants in the genome that differentiate people are of ancient human origin and occur commonly within and across populations. Nevertheless, every individual still possesses about 25,000-50,000 rare single nucleotide variants, in addition to about ten times as many small insertion/deletion variants. These “private” variants may be unique to any particular person and are shared only with their family members. When comparing people with ancestral origins from different parts of the world, there is actually greater genetic diversity within a local population than when comparing across geographically distant populations. Because most human history transpired before the migration out of Africa, most, but not all, genetic variants arose before that time and are distributed across people of all ancestries.
Genome – The complete sequence of DNA present in a cell or organism. The haploid human genome is ~3.3 GB (billion base pairs).
Exome – The protein-coding portion of the genome, which constitutes ~1% of the human genome or ~30 Mb (million base pairs, “megabases”), spread out across ~180,000 exons from ~20,000 total genes. Since most Mendelian disorders (but not complex diseases) are the result of mutations within the protein-coding portion of genes, clinical genetic testing for challenging diagnostic questions often relies on exome sequencing, as opposed to genome sequencing. Exome sequencing has several advantages: It is less costly. It returns less inconsequential data, including polymorphisms, requiring analysis. Based on the “next-generation” massively parallel technologies in use today, any given nucleotide is read multiple times, assuring greater accuracy than can be achieved with whole genome sequencing, where breadth of coverage occurs at the expense of depth of coverage. The high depth of coverage can be exploited to help identify potential large-scale deletions or insertions (“copy number changes”).
Sources of genetic variation. Although exposure to environmental mutagens, such as ionizing radiation, can alter DNA sequences, most changes occur spontaneously. Since most cells are “somatic” and do not contribute to gamete formation, these DNA sequence changes are not passed down from one generation to the next—although they may contribute to cancer. DNA changes occurring in germ cell precursors giving rise to eggs and sperm do, however, result in heritable changes to the “germline.”
The most common type of SNP in both somatic cells and in the germline is a change of a C:G base pair to a T:A base pair and reflects the fact that methylcytidine, an epigenetic mark occurring in CG sequences (usually referred to historically as “CpG” with the “p” representing the phosphodiester bond), spontaneously hydrolytically deaminates to thymine. Another common type of spontaneous mutation reflects oxidative damage to DNA. DNA damage occurs tens-to-hundreds of thousands of times per cell per day. Most damage is repaired by specific DNA damage response pathways, but the process is not completely perfect, resulting in a mutation. Many different mutational signatures can be linked to particular environmental carcinogens (e.g., tobacco smoke) or cell endogenous processes, such as noted for oxidative damage, making it possible in some cases to link particular environmental agents with the types of cancer they contribute to.
Once a germline DNA variant arises in any given individual, the extent to which it is propagated further among that person’s descendants depends upon both random events as well as the population’s size and natural selection if the variant has functional consequences. The random sampling of gametes during sexual reproduction leads to “genetic drift,” which is a stochastically introduced fluctuation in the population frequency of a genetic variant. When a population size is reduced (goes through a “bottleneck”), particular genetic variants may be subsequently over-represented through “founder effects,” merely because a modern population can be traced back to a small number of founding individuals. Finally, some DNA sequence changes alter gene activity and produce new phenotypes that may be advantageous or deleterious and are therefore subject to natural selection.
SLO 3 Explain how natural selection influences disease gene frequencies
Genetic “fitness” refers to the relative reproductive success imparted by a particular genetic variant. For example, a mutation producing a severe phenotype, such as an autosomal dominant Mendelian disease resulting in fatality during childhood, has zero reproductive fitness. Fitness also reflects external factors, such as the environment.
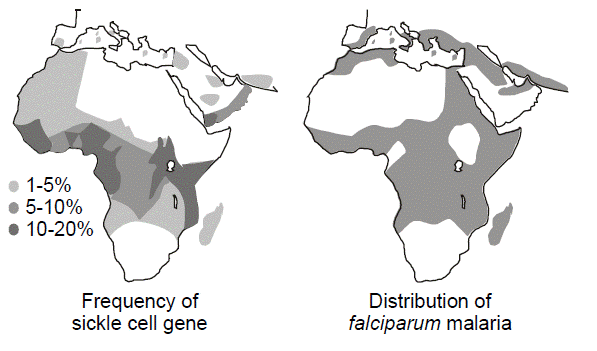 Heterozygote advantage. Selective pressures operate differently for autosomal recessive diseases than they do for dominant diseases. A low fitness for a recessive disease has much less effect on removing a disease allele from the population than it would for a dominant disease. This is because most of the alleles will exist within a reservoir of unaffected heterozygotes. On the other hand, selective pressures can influence the heterozygote population, which is large in comparison to the homozygous affected population. If the heterozygotes have a selective advantage compared to those who are homozygous for normal alleles, then the frequency of the disease allele will increase. Since heterozygotes are far more common than affected homozygotes, for any given disease allele, the benefit to the relatively large heterozygote population may be more than enough to make up for any deleterious effect upon fitness in the comparatively smaller homozygous population. This is the reason why sickle cell anemia and other hemoglobinopathies as well as thalassemia are so common in regions of the world where malaria is endemic. Sickle cell anemia is an autosomal recessive disease caused by an amino acid missense substitution in the beta chain of hemoglobin (designated Hb S). Heterozygous carriers of the sickle cell trait are relatively resistant to Plasmodium falciparum malarial infection and its complications. A similar heterozygote advantage is thought to explain the maintenance of recessive alleles for cystic fibrosis in the population. There is evidence that individuals heterozygous for cystic fibrosis mutations are relatively resistant to cholera and Salmonella enteritis.
Heterozygote advantage. Selective pressures operate differently for autosomal recessive diseases than they do for dominant diseases. A low fitness for a recessive disease has much less effect on removing a disease allele from the population than it would for a dominant disease. This is because most of the alleles will exist within a reservoir of unaffected heterozygotes. On the other hand, selective pressures can influence the heterozygote population, which is large in comparison to the homozygous affected population. If the heterozygotes have a selective advantage compared to those who are homozygous for normal alleles, then the frequency of the disease allele will increase. Since heterozygotes are far more common than affected homozygotes, for any given disease allele, the benefit to the relatively large heterozygote population may be more than enough to make up for any deleterious effect upon fitness in the comparatively smaller homozygous population. This is the reason why sickle cell anemia and other hemoglobinopathies as well as thalassemia are so common in regions of the world where malaria is endemic. Sickle cell anemia is an autosomal recessive disease caused by an amino acid missense substitution in the beta chain of hemoglobin (designated Hb S). Heterozygous carriers of the sickle cell trait are relatively resistant to Plasmodium falciparum malarial infection and its complications. A similar heterozygote advantage is thought to explain the maintenance of recessive alleles for cystic fibrosis in the population. There is evidence that individuals heterozygous for cystic fibrosis mutations are relatively resistant to cholera and Salmonella enteritis.
From the Hardy-Weinberg law, the heterozygote advantage results because 2pq >> q2, and explains the high carrier frequency for certain autosomal recessive disease alleles that confer disease resistance in a heterozygote state.
Carrier selection also occurs for sex-linked recessive disease where unaffected carriers are similarly more abundant than affected individuals, as in the case of glucose-6-phosphate dehydrogenase (G6PD) deficiency, which follows a geographic distribution similar to that of sickle cell disease and appears to confer similar resistance to malarial infection and its complications. G6PD participates in the hexose monophosphate shunt of glycolysis. In fact, since both Lyonization (X inactivation) and carrier selection apply in the case of G6PD deficiency, one can make a striking observation in microscopic examination of the peripheral blood smear of heterozygous female carriers infected with malaria. If the smear is stained to histochemically detect G6PD, then Lyonization will be discernable at the level of single cells—some erythrocytes will express G6PD and others won’t. Interestingly, the malarial parasites will be seen preferentially in the red blood cells that express G6PD and be relatively scarce in red blood cells that lack G6PD activity. A deficiency of G6PD activity results in hemolytic anemia upon exposure to oxidants, including sulfa antibiotics, quinine-based antimalarials, and fava beans. Ironically, problems with G6PD first became widely recognized when GI’s of African ancestry serving in Korea in the 1950s suffered disproportionately compared to individuals of other ancestries from side-effects of therapeutic treatment with prophylactic antimalarial drugs.
SLO 4 Explain how meiosis increases genetic diversity
In order to understand complex genetics, one must also understand the concept of genetic linkage. And to understand linkage, one must know about meiosis. Of course, meiosis is central to other concepts in genetics, such as the origins of constitutional chromosomal imbalances, which we will cover later.
Meiosis – is the cell division process in which “haploid” gametes are formed from “diploid” germ cells. Meiosis addresses the problem of preventing genome size from doubling at each generation in a sexual organism. Meiosis increases genetic diversity by randomly re-sorting chromosomes from the parents, the male making sperm and the female making eggs (i.e., the grandparents of the future conception). It is also the time when “recombination” between each parental chromosome homolog takes place, further increasing genetic diversity, by recombining the chromosomes themselves such that they become composites of each set of grandparents. Mistakes in meiosis are responsible for major chromosomal abnormalities.
Meiosis is fundamental to sex. Sex increases opportunity for genetic diversity, and therefore makes it more likely that at least some individuals within a population will be able to survive in a changing environment.
Meiosis is divided into two sequential stages, meiosis I and meiosis II. Meiosis I and meiosis II are then each subdivided into several substages that share names with each other and with the substages of somatic cell division (“mitosis”).
Meiosis accomplishes two things. The first is to prevent a doubling in the quantity of chromosomes from one generation to the next.
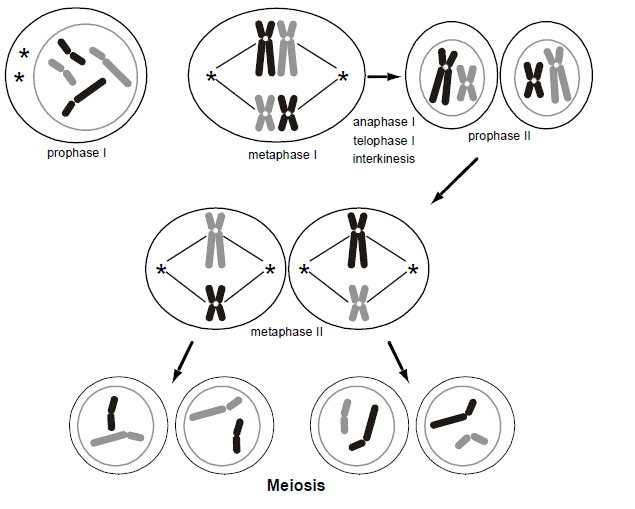
Normally, for every pair of chromosomes in every individual, one chromosome came from the mother’s egg and the other from the father’s sperm. During the longest part of the cell cycle (interphase), the chromosomes are not microscopically visible in a cell. Only during cell division and gametogenesis do the chromosomes condense enough to become microscopically visible (predominantly during the metaphases). The condensation facilitates their mitotic or meiotic segregation. During the formation of an egg or a sperm, one parental chromosome from each pair is randomly sorted for inclusion in the gamete. The way this is accomplished is that the 23 pairs of chromosomes form a physical pair (called a “synapse”) and then line up along the midline of the cell. In this figure, for simplicity, we are only showing two different chromosomes, distinguishable by their size. We are shading each chromosome, black vs. gray, to distinguish maternal from paternal contributions. Each pair of chromosomes lines up in a random orientation (referred to as “independent assortment”), so that in one pair the maternally inherited chromosome might be on the left and the paternally inherited chromosome on the right, while in another pair the opposite may be true. Then, the cell divides into two, so that only one member of the pair sorts into each of the two daughter cells. Consequently, there are a very large number of ways that the parental chromosome homologs can be distributed into each gamete, even in the absence of chromosomal recombination. This is known as meiosis I, and at this point there are now just 23 chromosomes per cell.
The chromosomes had completed a round of DNA replication, similar to mitosis, before entering into meiosis I. Each chromosome in the synaptic complex therefore has two “sister chromatid” arms that are side-by-side. Since there is a pair of “homologous” chromosomes in each synapse, each with two sister chromatids, there are a total of four chromatids in the synapse. The result is that the two daughter cells produced at the conclusion of meiosis I have just 23 chromosomes (a haploid quantity of chromosomes) but each chromosome is present in a duplicated form. Each daughter cell must then undergo a second event (meiosis II, which is similar to meiosis I except that there are only 23 instead of 46 chromosomes). During meiosis II, the sister chromatids are pulled apart, and each of the 23, now non-duplicated chromosomes with just a single chromatid per each arm, goes into two more daughter cells. The result is that there are now four daughter cells, each with a haploid quantity of non-duplicated chromosomes.
In the formation of a sperm, one diploid cell begins meiosis and four haploid spermatozoa result from it. A significant difference during oogenesis in the female is that meiosis produces only one haploid oocyte. One of the daughter cells resulting from meiosis I is discarded (and consequently does not enter meiosis II), and one of the daughter cells resulting from meiosis II is also discarded. These discarded cells are known as “polar bodies.” Rarely, a sperm aberrantly fertilizes a polar body, resulting in a hydatidiform mole which can evolve into choriocarcinoma, the only type of human cancer where the tumor genome is not that of the individual in whom it arises, but in this case is derived from the partner’s sperm.
Another significant way that meiosis differs between the sexes is that for males, meiosis is a continuous activity that begins at puberty and proceeds until death. For females, there are several thousand primitive oocytes arising in the developing ovary of a female embryo and that initiate meiosis I well before birth, but then arrest during embryonic development. Meiosis does not resume until after puberty, and then in only one egg at a time (with the egg that is ovulated during a particular menstrual cycle, at which point meiosis I is completed). Meiosis then again arrests, and meiosis II is not completed until just after fertilization, at which point the second polar body is ejected.
Recombination. The second accomplishment of meiosis is to produce genetically variable gametes through independent assortment (as noted above) and recombination. During genetic recombination, an individual’s two parental chromosome homologs actually physically break and recombine to produce a recombinant chromosome, containing combinations of grandparental alleles that were not previously linked in either parent. Recombination occurs in only two of the four chromatids present in the synapse. So, from every meiotic event there are always two potential recombinant chromosomes (that represent reciprocal exchanges) and two non-recombinant chromosomes.
above) and recombination. During genetic recombination, an individual’s two parental chromosome homologs actually physically break and recombine to produce a recombinant chromosome, containing combinations of grandparental alleles that were not previously linked in either parent. Recombination occurs in only two of the four chromatids present in the synapse. So, from every meiotic event there are always two potential recombinant chromosomes (that represent reciprocal exchanges) and two non-recombinant chromosomes.
SLO 5 Explain the concept of linkage disequilibrium and haplotypes
Genes are not always inherited independently from one another. The circumstance in which genes are independently inherited from one another is when they reside on different chromosomes. In this case, they are not physically “linked” to one another, because each chromosome is composed of a different linear molecule of DNA, and each pair of chromosome sorts independently from other pairs of chromosomes during meiosis. However, genes on the same chromosome are physically linked to one another. If there were no recombination, then genes residing on the same chromosome would always co-segregate with each other through meiosis.
Nevertheless, as explained above, recombination does occur between two parental chromosome homologs, for any particular chromosome, during meiosis. The consequence is that alleles for two physically linked genes on the same chromosome inherited from the same parent can be dissociated from one another in meiosis.
Recombination more or less occurs randomly across a chromosome. In general, at least one recombination event (a so-called “crossing over” event) occurs per chromosome arm per meiosis. Typically, there are several crossing overs per chromosome arm, involving combinations of any two of the four sister chromatids participating in the meiotic synapse.
In general, the further apart two genes on the same chromosome are from one another, then the more likely it is that there will be a recombination event occurring between them; consequently, genes distant from one another along the chromosome arm less often co-segregate with one another. Conversely, if two genes are close together, then it is much less likely that a crossing over event will occur between them. In fact, when two genes are adjacent to one another there is seldom recombination between them, and the two genes consistently co-segregate with one another. Nearby genes that tend to co-segregate with one another are referred to as being in “linkage disequilibrium.”
It is not just genes that demonstrate linkage disequilibrium. Any discernible genetic change, including a SNP (the different versions of which are also still referred to as “alleles”) can exhibit this property. Closely adjacent clusters of genes or other genomic markers or landmarks, including SNPs, tending to co-segregate with one another on the same parental chromosome, are referred to as a “haplotype.”
Linkage disequilibrium – alleles for genes or genomic landmarks, such as a SNP, close to one another (in physical proximity on the same chromosome) tend to co-segregate through families with one another, since it is unlikely that a recombination event will occur between them.
Haplotype – refers to a group of alleles from closely linked loci that are usually inherited together as a unit and that exhibit linkage disequilibrium.
Ultimately, recombination is a biochemical process involving DNA and proteins that can bind to DNA and physically recombine double-stranded molecules. Consequently, over time, recombination will whittle down the length of a haplotype but it most likely reaches a minimal size limit, in length defined by base pairs of DNA, that is both a consequence of the number of generations that have elapsed since the initial haplotype was defined, as well as the physical limits of recombination.
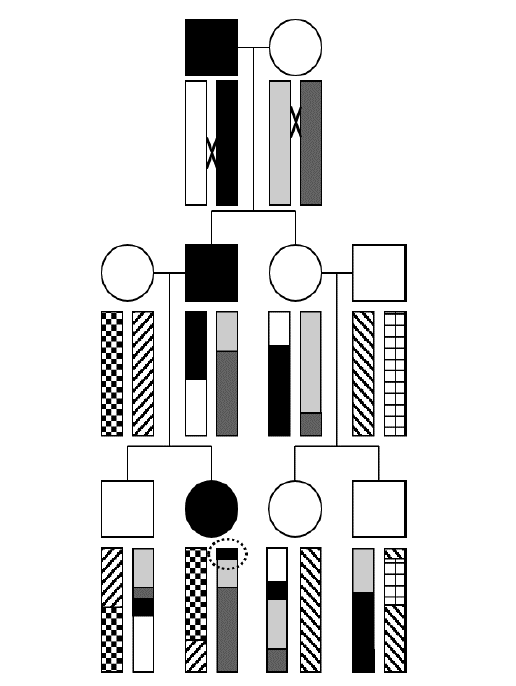
To illustrate the concept of how haplotypes, which are blocks of linkage disequilibrium, are created by recombination during meiosis, let’s examine this three-generation pedigree transmitting a highly penetrant autosomal dominant trait, where the affected individuals are filled-in with black.
The rectangles represent a chromosome arm. Note that each “ancestral” haplotype (which in this case corresponds to the entire chromosome) is uniformly shaded or patterned. Each of the four people who “marries into” the family introduces a new, fresh, ancestral haplotype, denoted with a unique pattern.
The “×” symbol indicates a site of recombination (or crossing over) in the first generation. Notice that the black and white ancestral haplotypes physically recombine in the affected male in the first generation and produce a new recombinant haplotype where the ancestral segment containing the causative mutation is shortened and inherited by the affected male child. His female partner also has a recombination event, and the chromosome containing a mixture of ancestral haplotypes is similarly inherited by their affected child. As the second generation choose mates of their own, new ancestral haplotypes are introduced, here represented by unbroken patterns. Recombination continues, and each ancestral haplotype is further reduced in size with each passing generation.
Can you determine the position on the ancestral chromosome where the responsible gene must reside? Hint: it’s circled! As you can see, that is the shortest haplotype inherited by all three affected individuals, and none of the unaffected individuals possess this haplotype at that region of the chromosome.
Linkage disequilibrium can be used to date the emergence of an allele in the population. For example, consider the CCR5-Δ32 allele, which affords resistance to human immunodeficiency virus (HIV) infection. CCR5 is a chemokine receptor protein present on the surface of macrophages. The Δ32 mutation deletes 32 nucleotides and creates a frameshift that prematurely leads to termination of the altered reading frame. Homozygotes for the Δ32 allele of CCR5 are advantageously resistant to infection by HIV. Heterozygotes appear to have intermediate levels of resistance to HIV. Chromosomes that have the Δ32 mutation on them all have the exact same alleles for various markers that flank the CCR5 gene extending on a small stretch on either side. From this haplotype sharing it can be deduced that everyone possessing this allele must be descended from a common ancestor. From theoretical studies it can be inferred that the length of the haplotype held in common between these modern-day individuals must have taken about 30 or so generations to reach this shortened length through progressive meiotic recombination events. It can thus be deduced that this mutation first arose about 700 years ago (assuming something like 23 years/generation) and is hypothesized to have coincided with the Plague in Europe. It would appear that this mutation was a relatively random event existing in one or a few related individuals in the Middle Ages and presumably became more common in the population because it conferred some degree of resistance to the Plague and quite incidentally, also HIV, which probably had not yet infected humans at that time. Yersinia pestis, the Plague bacillus, utilizes the CCR5 molecule to infect macrophages, as does HIV, explaining why mutant CCR5 would confer resistance to both pathogens.
Genetic Linkage Analysis. The above diagram showing co-segregation of a progressively shortening haplotype with a disease phenotype within a family actually depicts the process of “genetic linkage analysis,” which was the historical approach used to identify genes responsible for most Mendelian disorders. However, this was during a time before the Human Genome Project determined a reference genome sequence and before the current powerful “next-generation” of massively parallel DNA sequence technologies responsible for economical exome and genome sequencing came into being.
Today there are more powerful and less expensive technologies, namely, ready clinical access to whole exome and whole genome sequencing.
While most single-gene Mendelian disorders have already been identified, there are still a few that continue to be discovered because they are either rare or the spectrum of clinical findings associated with a known syndrome had not been fully appreciated previously. The ability to perform exome or genome sequencing on patients who have defied clinical diagnosis has brought fresh hope to patients and families with heretofore seemingly unexplained diseases.
SLO 6 Describe the origins of the haplotype block structure of the genome
Let’s return to the above figure demonstrating how a haplotype progressively shortens as a result of meiotic recombination with each passing generation. Instead of just observing this phenomenon for three generations, as in the figure, imagine what would happen over a great number of generations, say as many generations as have elapsed from the time when humans first migrated out of Africa and began populating the rest of the world. The haplotype continues to shorten, but it does not become infinitesimally small and instead approaches a minimal limit, probably ultimately restricted in size by the biochemical limits imposed by the recombination process itself.
Therefore, each of our chromosomes is a mosaic of ancient, ancestral haplotypes, averaging from tens of thousands to hundreds of thousands of base pairs in length. These ancient haplotype blocks are distinguishable by the unique variants, mostly SNPs, but also insertions and deletions and other types of polymorphisms, that arose long ago and that define the ancestral haplotype.
And since populations tend to grow exponentially, modern populations are descended from relatively few founders. Therefore, the set of chromosomes amongst all people in contemporary populations achieve their genetic diversity by mixing and matching “blocks” representing a relatively small number of ancient haplotypes. And within a haplotype block, adjacent polymorphisms are all in linkage disequilibrium. That is, they continue to segregate with one another in a pedigree. In many cases, determining the genotype of just one SNP at a particular location in the human genome is sufficient to uniquely identify the particular ancestral block and infer adjacent sequence along the chromosome for a few hundred thousand base pairs in either direction. Of course, new mutations and polymorphisms have sprung up over time within an ancient haplotype block, but, in general, these are relatively few and far between; when comparing any particular ancient haplotype block shared by any two people possessing that particular block, the similarities in DNA sequence will be far greater than the differences.
The international scientific collaboration that has mapped the haplotype structure of the human genome across populations has produced what has come to be known as the “HapMap.” The collection of SNPs that can define a particular haplotype block are sometimes referred to as “tag SNPs.” To a very good approximation, determining the genotypes for 300,000 to a million of these tag SNPs (one or a few per each block) can be used to stitch together the sequence of haplotype blocks and infer (or in technical jargon, “impute”) the entire genome sequence of any given individual, with the significant exception of polymorphisms and mutations that have arisen in any particular family in more recent generations.
Which particular haplotype blocks persist in the population has to do with chance (genetic drift), population bottlenecks, and any potential beneficial or deleterious influences that genetic variants contained within the block might confer (natural selection).
SLO 7 Explain how “genome-wide association analysis” (GWAS) is used to identify common genetic variants in the population that contribute to risk for common disease and how these variants differ from those responsible for Mendelian disorders
Let’s recap what we’ve learned so far about complex genetics:
- There is a significant underlying genetic component to common disease, distributed at multiple loci across the genome, each with weak effect.
- Most of the genetic variation in the population is ancient and therefore common, in that any particular variant is shared by a large number of people.
- There is a block-like structure to the human genome such that an individual’s genome represents a composite of a large but ultimately finite number of blocks measuring from a few tens or hundreds of thousands of nucleotides in length.
- Any given block is distinguished by a collection of SNPs unique to that block, so that if we determine the genotype of only one or maybe a few of these so-called tag SNPs we can, more or less, be confident of the DNA sequence of the entire haplotype block.
How can we put this all together to define the molecular genetic basis of common diseases? The answer is a “genome-wide association analysis” (GWAS).
Genome-wide association analysis (GWAS) – In a nutshell, a GWAS operates under the hypothesis that common genetic variants contribute to risk for common disease. GWAS consists of the following steps. A large number of cases and controls are assembled, with hundreds of thousands to millions of subjects typically now participating in any single GWAS. For each subject, cases and controls included, genotypes are determined for about 300,000 to a million or so tag SNPs dispersed across the genome. (Genotyping SNPs can be performed rapidly and economically using a variety of high throughput technologies.) Then a simple association analysis, using a chi-square statistical test, is employed to determine which SNPs are found more frequently or less frequently in cases compared to controls. Those SNPs occurring more frequently in cases are deduced to confer risk for disease, whereas those that occur more frequently in controls are considered protective.
Note that because each SNP only tags a haplotype block, genetic variation anywhere within that ancestral haplotype block may be responsible for the observed effect on disease risk. In other words, the tag SNP is chosen only for convenience, and the fact that it is associated with a particular disease does not necessarily imply that particular variant is causative. It could be another SNP or other polymorphism in linkage disequilibrium within that haplotype block or it could be a combination of polymorphisms acting in concert with one another. Similarly, the haplotype block may contain several genes. Once an association with a particular genetic region has been established it may not be possible to definitively identify what gene is contributing to the effect, and it is even more challenging to identify what particular genetic variant (or combination of variants) within a given gene is responsible.
GWAS has now been successfully used in many large studies to identify common variants (present in about 5-50% of individuals within a population) with small effect sizes (increasing risk by about 20-50%) for virtually every common disease (such as diabetes mellitus, coronary artery disease, chronic kidney disease, and autoimmune disorders like systemic lupus erythematosus), including for those with a quantitative component (for example, hypertension). The results can be biologically interesting. For instance, GWAS of lung cancer and chronic obstructive pulmonary disease (COPD) has—perhaps unsurprisingly—identified genetic variants in nicotinic receptors, such as the gene CHRNA5, suggesting that inborn genetic variation in vulnerability to tobacco addiction contributes to risk for developing smoking-related diseases.
Mendelian randomization. Recently, GWAS studies have been ingeniously repurposed to investigate how environmental factors influence disease risk. Mendelian randomization is a statistical method that uses genetic variants as natural experiments to infer causal relationships between modifiable risk factors (such as cholesterol levels or smoking) and health outcomes (like cardiovascular disease or cancer). This approach relies on the principle that alleles are randomly assorted during gamete formation and conception, mimicking the randomization process of a clinical trial in a way that is less likely than epidemiological studies to be affected by confounding factors or reverse causation (example of the latter: diet soda may incorrectly appear associated with obesity because people trying to lose weight avoid sugary soft drinks). If a genetic variant is reliably associated with a particular exposure, and that exposure is causally related to an outcome, then individuals carrying the variant should have different risk for the outcome in a way that reflects the effect of the exposure.
For example, Mendelian randomization has been used to demonstrate that elevated LDL cholesterol causally increases the risk of coronary artery disease. Individuals carrying variants in the PCSK9 or LDLR genes that lower LDL levels have reduced cardiovascular risk, supporting a causal relationship that is unlikely to be confounded by lifestyle or socioeconomic factors. In contrast, another Mendelian randomization study examined the CHRNA5 gene, associated with heavier smoking behavior, to assess the impact of smoking on lung cancer. Here, the environmental influence—smoking—is included and indirectly assessed via the genetic predisposition to smoke more. Because the gene affects disease only through its influence on smoking behavior, the analysis strengthens the evidence that smoking causally increases lung cancer risk, while reducing confounding from factors like occupational exposure or education level.
Consumer-level genetic testing. In addition to offering information about genetic ancestry, widely available, low-cost consumer-level genetic testing, such as through “23andMe,” reports genotype results for variants originally identified through GWAS along with genotype determination for some common alleles for Mendelian disorders, such as sickle cell disease and common mutations of CFTR. They also report findings for other common disorders that blur the boundary between complex disease and single gene Mendelian disorders, such as late-onset Alzheimer disease resulting from the APOE E4 allele, which behaves as a Mendelian disorder but where the disease-associated allele is very common in the general population.
GWAS results are frequently misinterpreted by patients and physicians alike. In general, Mendelian disorders are uncommon, highly penetrant, and the responsible mutations are restricted to individual families and fairly obviously disrupt the protein-coding sequence of a gene. In contrast, variants contributing to common disorders confer only incremental risk and act additively with one another in concert with lifestyle and environmental exposures and their effect on how a gene functions may be difficult to tease apart.
The first human genome sequence was completed in 2003, required thousands of scientists working in laboratories throughout the world, and cost nearly $3,000,000,000. Direct-to-consumer whole genome sequencing now costs under $300. One can only begin to imagine how this information will revolutionize the practice of medicine and strain the ability of patients and health care providers to make sense of the data.
Feedback: