
7 Chromosomal abnormalities, Aneuploidies, Chromosomal Rearrangements
Session Learning Objectives and a quick synopsis:
Genes reside on chromosomes. Chromosomes can break, lose or gain material, rearrange, and become lost or duplicated. Chromosomal abnormalities disrupt many genes at once. Some chromosomal abnormalities are restricted to the person in which they are found. Some chromosomal abnormalities can be inherited. Some chromosomal abnormalities can be inherited from a “balanced carrier,” in which the parent has structurally abnormal chromosomes that have neither lost or gained genetic material but can then rearrange further during meiosis (formation of a sperm or egg) in a way that causes loss and/or gain of genetic material to become “unbalanced”.
SLO 1 Describe the cytogenetic organization of the human genome
The diploid human genome consists of about six billion base pairs of DNA organized into 23 pairs of chromosomes, comprised of 22 pairs of autosomes plus an XX or XY pair of sex chromosomes.
A karyotype consists of a microscopic image of condensed metaphase chromosomes. Only large-scale copy number changes (interstitial duplications and deletions) and structural rearrangements can be visualized. It requires a source of living cells. Fluorescence in situ hybridization (FISH) is a different microscopic technique that has much higher resolution, can be performed on metaphase or interphase cells, and even fixed (preserved) cells under certain circumstances, but is directed at probing for the integrity of certain chromosomal regions based on an underlying clinical hypothesis. Chromosome microarrays offer a high-resolution global view of the genome and only require a source of DNA rather than intact cells but are incapable of detecting balanced rearrangements. Karyotype and FISH are time and labor intensive, whereas microarrays can be performed in high throughput manner.
SLO 3 Describe human chromosomal abnormalities and how they are inherited
Some are inherited, some occur de novo in the germline, and others are restricted to somatic tissues, particularly cancers. Chromosome number imbalances are often the result of meiotic nondisjunction occurring as a function of maternal age, whereas structural rearrangements can occur via this or other mechanisms. The range of abnormalities includes loss or duplication of chromosomes; translocations; and interstitial insertions, deletions, and inversions. Chromosomal abnormalities disturb many genes, resulting in a severe clinical phenotype. An exception is sex chromosome abnormalities, which are clinically less pronounced. There are virtually unlimited numbers of possible ways that chromosomes can rearrange, meaning that many patients have unique and previously unseen clinical phenotypes. Nevertheless, there are recurrent rearrangements in which patients share common clinical features. Recurrent rearrangements seen in some cancers have diagnostic, prognostic, and treatment significance. Some chromosomal abnormalities result in no clinical abnormalities in parents but can promote deleterious rearrangements occurring during meiosis. Different diagnostic technologies are required for different types of changes, as noted in SLO2.
SLO 4 Distinguish constitutional from acquired chromosomal abnormalities
Constitutional cytogenetic abnormality: chromosome make-up at birth that may be heritable. Acquired cytogenetic abnormality: typically not present at birth, limited to certain tissues, and not heritable.
SLO 5 List clinical scenarios for which a chromosomal abnormality should be considered
Indications for suspecting a chromosome abnormality include multiple congenital malformations, intellectual disability, and growth failure, an overall clinical picture that resembles a specific known syndrome involving a chromosomal abnormality, recurrent pregnancy loss, infertility, short stature or primary amenorrhea, ambiguous genitalia, and hematologic malignancies and certain solid tumors.
Main text
1/150 newborns have a chromosome abnormality. At least 50% of first trimester spontaneous abortions (“miscarriages”) have a chromosome abnormality. Approximately 2% of all pregnancies in women over 35 years-old have a chromosomal abnormality. Acquired chromosomal abnormalities are a common feature of malignancies.
SLO 1 Describe the cytogenetic organization of the human genome
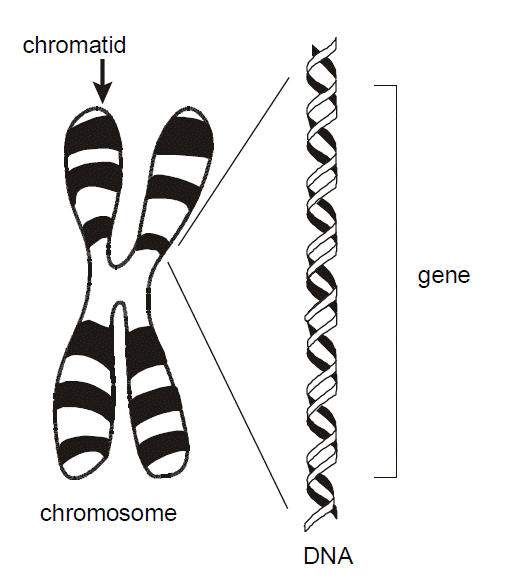
To review, the diploid human genome consists of about six billion base pairs of DNA organized into 23 pairs of chromosomes. Each chromosome consists of a single linear continuous strand of DNA complexed with histone and other proteins. This combination of DNA and protein is arranged in structures of progressively greater degrees of organization. The double helix is wound into a nucleosome fiber, that in turn is coiled into a solenoid, then assembled into chromatin, and finally the chromatid.
Chromosome – A single DNA molecule condensed on a protein scaffold in the nucleus of a cell. Each chromosome contains hundreds or thousands of genes. Humans have 22 pairs of “autosomes” and one pair of sex chromosomes (X and Y), for a total of 46.
Chromatid – One of two identical parallel DNA strands in a mitotic cell following DNA replication.
Why do we need chromosomes? Although only one twenty-millionth of a meter-wide, the extended length of the human genome would reach several meters unless there were a means for compactly folding and organizing the DNA to fit within the confines of a cell.
Cytogenetics. Cytogenetics refers to the study of chromosomes, their structure, and their inheritance.
SLO 2 Compare and contrast the clinical laboratory tests (karyotype, FISH, and microarrays) used for detecting chromosomal abnormalities
 Karyotype. Microscopic staining and visualization of chromosomes (a “karyotype”) is usually performed on dividing cells. Peripheral blood leukocytes are most often used for routine chromosome analysis because of the ease of obtaining the specimen. Other tissues used for chromosome analysis include bone marrow samples (used mainly for the diagnosis of hematologic malignancies), solid tumors, skin (used frequently to detect mosaicism), and amniotic fluid cells and chorionic villi (for prenatal diagnosis of chromosomal disorders). The cells are grown in culture for a short time, then treated with the microtubule polymerization inhibitor colchicine to arrest the cells during the metaphase of mitosis, when chromosomes are maximally condensed. The cells are lysed, the chromosomes are fixed to a glass slide, then stained. Photomicrographs are obtained and images of individual chromosomes are cut out and pasted together for comparison.
Karyotype. Microscopic staining and visualization of chromosomes (a “karyotype”) is usually performed on dividing cells. Peripheral blood leukocytes are most often used for routine chromosome analysis because of the ease of obtaining the specimen. Other tissues used for chromosome analysis include bone marrow samples (used mainly for the diagnosis of hematologic malignancies), solid tumors, skin (used frequently to detect mosaicism), and amniotic fluid cells and chorionic villi (for prenatal diagnosis of chromosomal disorders). The cells are grown in culture for a short time, then treated with the microtubule polymerization inhibitor colchicine to arrest the cells during the metaphase of mitosis, when chromosomes are maximally condensed. The cells are lysed, the chromosomes are fixed to a glass slide, then stained. Photomicrographs are obtained and images of individual chromosomes are cut out and pasted together for comparison.
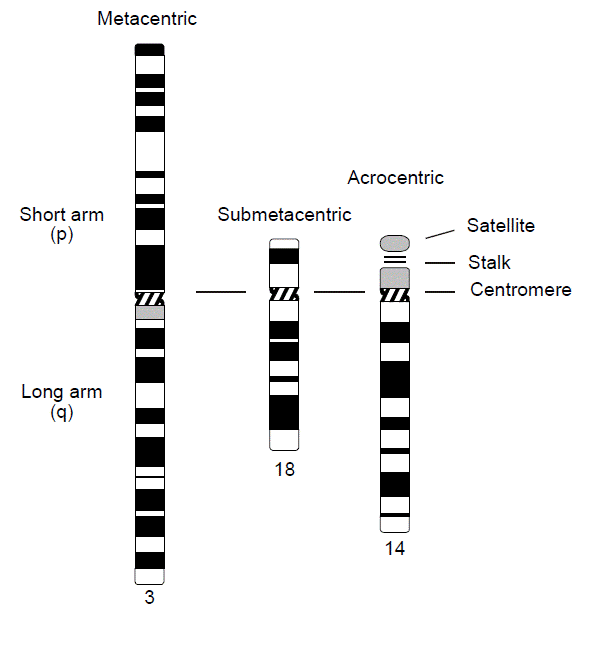
Chromosomes are classified into different groups on the basis of centromere position and chromosome size. The centromere is a standard cytological landmark that divides the chromosome into two arms—p (for petite), or short arm, and q (because it’s the next letter of the alphabet) for the long arm. Human chromosomes are classified by centromere position into three types—metacentric (arms of approximately equal length), submetacentric (arms of unequal length), and acrocentric (centromere near one end of the chromosome). The p arm of the acrocentric chromosome is composed of a satellite and stalk region that contains copies of ribosomal RNA genes. Because there are a half-dozen acrocentric chromosomes, ribosomal RNA genes exist in vast redundancy, so deletion of a p arm from an acrocentric chromosome (mainly through chromosome translocation) is usually not clinically meaningful.
 In addition, the chromosomes are numbered and arranged according to size, from largest to smallest, and position of their centromere, except for the sex chromosomes, which are always placed in the lower right-hand corner. This ordered configuration of chromosomes is called a karyotype. Note that chromosome numbers were assigned in the era before DNA sequencing became available. While they are supposed to be numbered from longest to shortest, we now know that chromosome 21 is actually shorter than chromosome 22.
In addition, the chromosomes are numbered and arranged according to size, from largest to smallest, and position of their centromere, except for the sex chromosomes, which are always placed in the lower right-hand corner. This ordered configuration of chromosomes is called a karyotype. Note that chromosome numbers were assigned in the era before DNA sequencing became available. While they are supposed to be numbered from longest to shortest, we now know that chromosome 21 is actually shorter than chromosome 22.
Karyotype – Microphotographic array of stained chromosomes ordered according to their length and the relative position of their centromeres. Cells to be karyotyped must be obtained when they are still living and capable of dividing. Karyotypes are not capable of resolving chromosomal abnormalities smaller than a few million base pairs in length.
Each chromosome has a characteristic banding pattern when stained. Cytogenetic analysis in the laboratory can produce various levels of resolution based upon the quality and origin of the chromosome preparation. Typically, the greater the number of bands that can be distinguished, the better the resolution.
An idealized drawing of the human karyotype is referred to as an “ideogram.” The ideogram shown here represents a resolution of 400 bands. Notice that the bands have been numbered with a standardized system, which allows one to define structural and numerical abnormalities. Each arm is first divided into regions (outer numbers) and then each region into specific bands (inner numbers). Numbering is from the centromere outward on each arm, the region number, and finally the band number. For example, band 2p21 (pronounced “two-pee-two-one”) indicates chromosome 2, short arm, region 2, band 1.
A standardized system of karyotype nomenclature has been defined. The system of notation is as follows: first indicate the total number of chromosomes, followed by a comma and then by the chromosome constitution. For example, 46,XX for female and 46,XY for male. If an abnormality is present, it is described using specific notations. For example, 47,XY,+21 (male with trisomy 21 Down syndrome).
Routine and high-resolution chromosome analysis permit the detection of numerical and structural chromosome abnormalities, but only to the extent to which we can actually see these changes under the microscope. Small chromosomal deletions, translocations and “marker” (fragmentary) chromosomes are difficult and sometimes impossible to identify with routine analysis. Even under the best conditions, a karyotype cannot resolve chromosomal changes smaller than 5-15 million base pairs.
Fluorescence in situ hybridization (FISH). Fluorescence in situ hybridization (FISH) provides the ability to simultaneously assess molecular and cytologic information. FISH analysis also allows for the identification of small deletions, translocations, and other chromosomal rearrangements too small to be detectable by ordinary karyotype. Various FISH protocols can distinguish chromosomal abnormalities as small as a few thousand base pairs. Another distinct advantage of FISH is that it can be performed on interphase cells, when the chromosomes are maximally unwound and extended. Unlike with a karyotype where cells are arrested when chromosomes are condensed during mitosis, FISH can be performed without a round of cell division immediately upon obtaining a sample of blood or other tissue, and results can consequently be returned to patients more quickly.
FISH is a technique in which a labeled chromosome-specific DNA segment (probe) is hybridized with chromosomes and then visualized under a fluorescence microscope. Several types of FISH probes are available depending upon the application in question. Probes to sequences repeated in the centromere and that are unique to each chromosome produce intense, tight signals that are easy to count in interphase nuclei and can therefore be used to quickly differentiate certain trisomies, such as trisomy 21 Down syndrome.
Microarrays. An approach for evaluating for small chromosomal abnormalities involves the use of microarrays. Two main platforms in use today are array comparative genomic hybridization (aCGH) and SNP arrays. The two approaches are similar and employ a “chip” onto which thousands of oligonucleotides are individually immobilized onto spots in an array. Genomic DNA is then hybridized to the array. If there is a deletion, then there will be an absence of binding to the spotted oligonucleotides corresponding to that position in the genome. If there is a genomic duplication, then there will be excess binding to the corresponding positions. Microarrays have several advantages compared to karyotype analysis and even FISH. Unlike karyotype and FISH, a microscope is not incorporated into the analysis. They therefore offer greater resolution. Purified DNA can also be analyzed whereas karyotypes and FISH require intact chromosomes typically obtained from growing cells. There are advantages in cost and throughput, as well. One weakness with microarrays is that they cannot detect “balanced” rearrangements, such as translocations or inversions, where the linear arrangement and relative chromosomal placement of genes are disturbed but the overall content of the genome otherwise remains intact.
SLO 3 Describe human chromosomal abnormalities and how they are inherited
Aneuploidy – refers to an extra or missing chromosome. It is a common type of numerical chromosomal abnormality and can involve either autosomes or sex chromosomes. Trisomy 21 Down syndrome is an example of an autosomal aneuploidy. 47,XXY Klinefelter syndrome and 45,X Turner syndrome are examples of sex chromosome aneuploidies.
Chromosomal abnormalities may be divided into two broad categories—numerical and structural abnormalities. Numerical abnormalities are simply abnormalities of chromosome number, typically involving just one chromosome (i.e., “aneuploidy,” such as trisomy 21 Down syndrome). Structural abnormalities include chromosomal rearrangements, deletions, and duplications.
Numerical abnormalities. Numerical abnormalities consist of those due to extra or missing chromosomes (aneuploidy) as well as multiples of the haploid chromosome number (polyploidy). Aneuploidy may involve either autosomes or sex chromosomes. Most common are monosomies (45 chromosomes) and trisomies (47 chromosomes).
Most aneuploid conditions are the result of nondisjunction, the failure of chromosomes engaged in a synaptic complex to separate during meiosis. At the top of the figure, each parental homolog of the same chromosome is depicted in a synaptic complex. Note that nondisjunction can occur at either meiosis I or meiosis II. When it occurs at meiosis I (left side of the figure), both parental homologs are included in the first division products, but they then segregate normally, except for the fact that there are two copies of one chromosome. Conversely (right side of figure), meiosis I can proceed normally, followed by nondisjunction in meiosis II. When that happens, two copies of the same parental homolog segregate into the gamete. In either case, however, following fertilization with a gamete containing a normal chromosome complement, the resulting zygote will either be trisomic or monosomic for the chromosome that did not disjoin. For simplicity, we are ignoring the impact of recombination in the nondisjunction process. In reality, the effect of 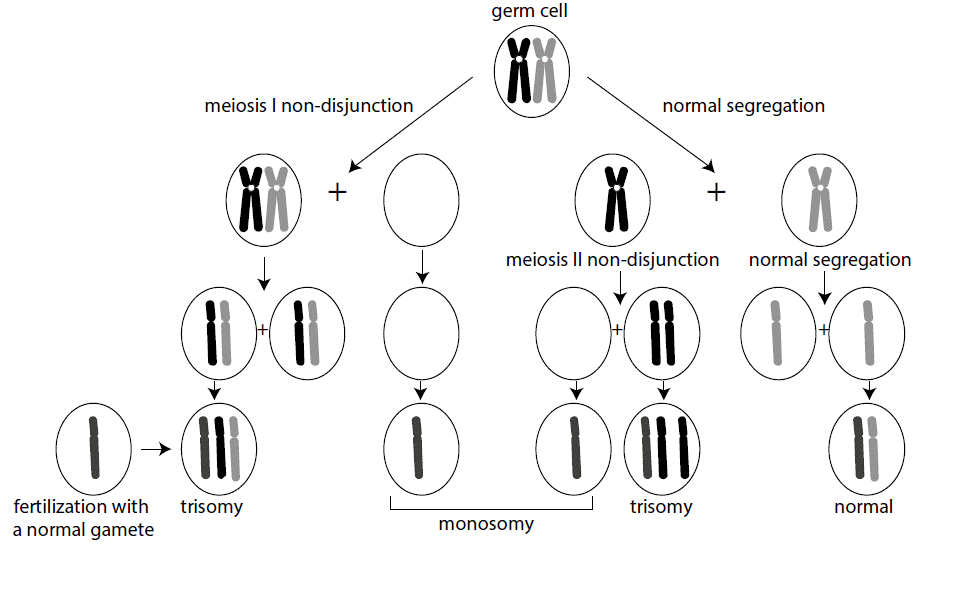 recombination can be used to determine whether the nondisjunction event occurred in meiosis I or meiosis II. From such studies, it has been found that the overwhelming majority of cases of nondisjunction occur during meiosis I of maternal gametogenesis.
recombination can be used to determine whether the nondisjunction event occurred in meiosis I or meiosis II. From such studies, it has been found that the overwhelming majority of cases of nondisjunction occur during meiosis I of maternal gametogenesis.
Most autosomal aneuploidy is the result of maternal meiosis I non-disjunction, which is associated with advanced maternal age.
Autosomal aneuploidy. There are three survivable trisomy syndromes that may result in a live birth, trisomy 21 (Down syndrome), trisomy 18 (Edwards syndrome), and trisomy 13 (Patau syndrome).
Down syndrome (Trisomy 21). The most common autosomal trisomy is Down syndrome, with an incidence of 1/800 live births. It is the most common cause of moderate intellectual disability. Individuals with Down syndrome have characteristic findings including hypotonia, unique features involving the face and head (flat nasal bridge, upslanting palpebral fissures, epicanthal folds, lightly colored “Brushfield” spots of the iris, flat midface, small dysplastic ears, protruding tongue, brachycephaly—“short” head, excess nuchal skin), hand (short and broad with fifth finger mid-phalanx hypoplasia and a single transverse palmar—“simian”—crease), congenital heart disease (most commonly endocardial cushion defects involving persistence of the atrioventricular canal), duodenal atresia, as well as an increased risk of developing leukemia, hypothyroidism, and early onset Alzheimer disease. Most individuals with Down syndrome have trisomy 21 due to nondisjunction. Maternal nondisjunction is typical (usually meiosis I error), and Down syndrome is associated with advanced maternal age (AMA).
It is probably no coincidence that trisomy 21 involves the shortest chromosome (therefore having among the fewest genes) and is the least severe of chromosomal trisomies; presumably, this is because fewer genes are triplicated compared to trisomies involving larger chromosomes.
In a minority of cases (approximately 2-4%), Down syndrome is the result of an unbalanced “Robertsonian” translocation leading to trisomy of the long arm of chromosome 21. A Robertsonian translocation is a special type of chromosome involving fusion of the long arms of two acrocentric chromosomes. In such cases, one parent is a Robertsonian translocation carrier, which predisposes to offspring with an unbalanced chromosome complement. The chromosomes may segregate abnormally in the offspring of a Robertsonian translocation carrier.
Even though the diagnosis of Down syndrome can be made fairly confidently based on the history and physical examination alone, it is important to always obtain a karyotype on a child with a clinical diagnosis of Down syndrome, both for confirmation and to determine if the child is the result of a Robertsonian translocation—in which case either parent could be a carrier and future pregnancies would be at risk for recurrence.
Characteristics of an autosomal chromosomal imbalance are the triad of developmental delay/intellectual disability, growth retardation, and multiple congenital abnormalities that often results in early fetal or neonatal demise. In contrast, sex chromosome aneuploidy tends toward comparatively mild phenotypes.
Uniparental disomy. As previously discussed in the session on other forms of inheritance, uniparental disomy (sometimes referred to as isodisomy) is the inheritance of both copies of a chromosome (or part of a chromosome) from just one parent, rather than the usual situation of inheriting one copy from each parent. Recall that uniparental disomy causes problems in two situations. First, if one parent is a heterozygous carrier for a recessive disease, then duplication of that chromosome will cause the conception to be homozygous even when the other parent is not a carrier. Second, if the individual has uniparental disomy for a chromosome or portion of a chromosome that includes an imprinted gene (such that it is not expressed from the parental sex corresponding to the duplicated (disomic) chromosome), then there will be loss of expression of that gene (because a chromosome from the other parent who would ordinarily express the imprinted gene is not present). Probably the most common mechanism for uniparental disomy is “rescue” of a conception that was initially trisomic. In this situation, the early embryo, probably consisting of no more than a few cells, is not-viable. However, if there is a mitotic non-disjunction event that kicks out one of the extra chromosomes, then that particular cell of the embryo may divide to clonally expand and give rise to a developing embryo. Note that in such a trisomic cell, either one of the duplicated chromosomes or the parental chromosome present in just a single copy can be lost. If it is the latter, then the result will be uniparental disomy.
Sex chromosome aneuploidy. Monosomy and trisomy of the sex chromosomes also occurs not uncommonly. The following are examples of sex chromosome abnormalities.
Turner syndrome (45,X). The incidence of Turner syndrome is approximately 1 in 5,000 live female births. Individuals with Turner syndrome, caused by near or complete absence of an X chromosome, have many features in common including short stature, lymphedema (resulting in a “puffy” appearance) of hands and feet at birth, webbed neck, broad chest with widely-spaced nipples, cubitus valgus (widened carrying angle of the arms), congenital heart disease (coarctation of aorta being most common), kidney abnormalities and gonadal dysgenesis resulting in a lack of secondary sex characteristics, and amenorrhea and infertility in the vast majority of females. Turner syndrome females have normal intelligence but may have difficulties with spatial perception.
Klinefelter syndrome (47,XXY). Males with Klinefelter syndrome have an additional X chromosome. The incidence of this sex chromosome abnormality is 1 in 1,000 live male births. These males tend to have tall stature, “eunuchoid” habitus, gynecomastia, and testicular atrophy. They typically do not have intellectual disability but may have some learning disorders. The extra X chromosome is maternal in origin in most cases, and there is an increased incidence of Klinefelter syndrome with advanced maternal age. Many of the clinical problems can be ameliorated with testosterone therapy. The extra X chromosome is typically inactivated and forms a Barr body.
Structural chromosomal abnormalities. Structural chromosome abnormalities may be balanced or unbalanced. By “balanced,” we mean that the rearrangements do not produce a net loss or gain of genomic material and usually cause no phenotypic effect. Rarely, the “breakpoints” for a structural chromosomal abnormality may disrupt a gene, but that is the exception, given that genes are relatively sparsely distributed across the entire genome. However, individuals with a balanced rearrangement have an increased risk for producing unbalanced gametes, which, in turn, if fertilized, produce conceptions with a complement of chromosomes containing partial deletions and/or duplications. Examples of balanced rearrangements include reciprocal and Robertsonian translocations (as noted, a specific type of translocation involving acrocentric chromosomes) as well as chromosomal inversions.
Translocations. Translocations involve the exchange of genetic material between nonhomologous chromosomal regions (or different regions, such as the long and short arms of the same chromosome).
Reciprocal chromosomal translocations. Reciprocal translocations involve the breakage of nonhomologous chromosomes with reciprocal exchange of chromosome material.
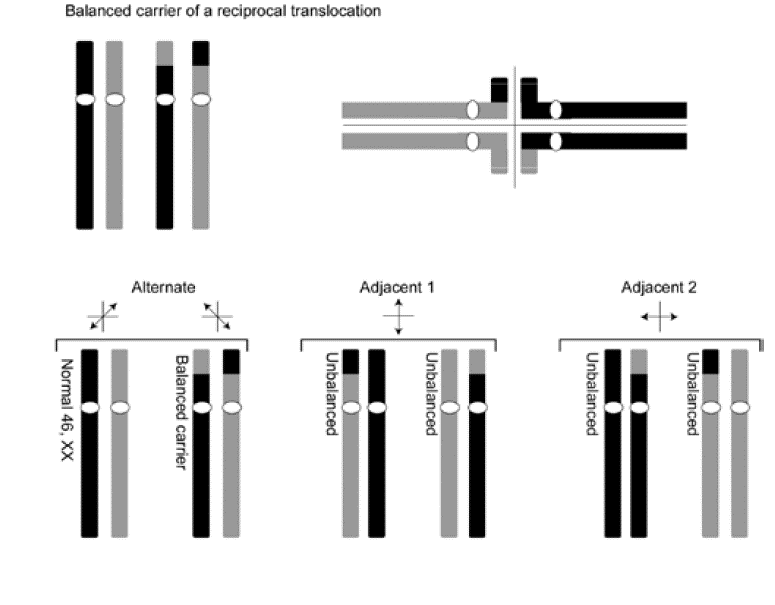 The carrier of a reciprocal translocation is usually phenotypically normal. However, carriers have an increased risk for producing unbalanced gametes and abnormal phenotype in their offspring.
The carrier of a reciprocal translocation is usually phenotypically normal. However, carriers have an increased risk for producing unbalanced gametes and abnormal phenotype in their offspring.
When chromosomes undergo translocation they have to form complicated structures to achieve paired synapses during meiosis. An example is shown of the meiotic segregation patterns that are possible in the carrier of a balanced, reciprocal translocation involving two chromosomes, colored black and gray and showing how they pair during meiosis. In this example, the parent carries a reciprocal translocation. Notice that the two translocation chromosomes form an unusual “quadriradial” synapse, which may be resolved in one of three ways. Alternate segregation yields one gamete with a normal complement of chromosomes and another gamete containing the pair of reciprocally balanced translocation chromosomes. Each of these gametes should produce an individual with a normal phenotype following fertilization by a normal gamete of the opposite sex. Either of the adjacent segregation patterns, however, yields unbalanced gametes. Each gamete contains a partial trisomy and a partial monosomy and is therefore expected upon fertilization to produce an abnormal phenotype.
It would appear that the risk of producing an abnormal offspring would be quite high (2/3 or 67%); however, due to prenatal loss of chromosomally abnormal offspring, the empiric risk of an unbalanced offspring from a parent carrying a balanced reciprocal translocation is much lower (approximately 5-15%).
Chromosomal inversions. Inversions within a chromosome result from two breaks on a chromosome with inversion of the segment and reinsertion at its original site. Approximately 1 in 1,000 individuals carries an inversion. Inversions are balanced structural rearrangements and therefore inversion carriers are usually phenotypically normal (no loss or gain of genetic material). However, during meiosis a chromosome with an inversion cannot normally pair with a structurally intact chromosome, and carriers are at risk to produce offspring with deletions or duplications and resultant abnormal phenotype. Inversions are classified as to whether they do or do not span the centromere because the two types behave differently during meiosis, with attendant implications for reproductive outcomes.
Chromosomal deletions. Deletions within a chromosome are caused by a break in the chromosome with resultant loss of chromosome material. Deletions result in an abnormal phenotype due to the loss of one or more genes.
A well-documented deletion syndrome is Cri-du-Chat syndrome (5p-), involving loss of the terminus of the short arm of chromosome 5. Infants with Cri-du-Chat syndrome have a distinctive “cat-like” cry as well as microcephaly, profound intellectual disability, growth retardation, and a unique facial appearance.
Some deletions, as well as duplications, are too small to see under the light microscope and hence go undiagnosed by karyotype. They are only detectable by molecular cytogenetic techniques such as FISH or microarrays. These conditions are referred to as “microdeletion” (or “microduplication,” “contiguous gene,” or “copy number variant (CNV)” disorders).
22q11.2 deletion syndrome (previously going by several different names including CATCH22 syndrome, DiGeorge syndrome, and velocardiofacial syndrome) is probably the most common microdeletion syndrome, with an estimated incidence of about 1/3,000 live births. It results from a deletion of the long arm of one chromosome 22 at band q11.2, spanning several megabases of DNA in which several dozen genes are located. Affected individuals may have cardiac abnormality, abnormal facies, thymic aplasia, cleft palate, hypocalcemia/hypoparathyroidism, and elevated risk for schizophrenia, in addition to other problems. Many of these problems can be tied together developmentally due to abnormal anatomic development of the fourth pharyngeal arch in the embryo. There is variability of the phenotype. 22q11.2 deletion often arises de novo in an affected individual whose parents are unaffected and who lack the causative chromosomal abnormality, but it can also be inherited in an autosomal dominant fashion, in which case affected individuals have a 50% chance of having a child with this disorder with each pregnancy.
One reason for variability in microdeletion syndromes is that different individuals will each have a deletion of varying extent sometimes involving different flanking genes. Nevertheless, a common collection of overlapping phenotypes, resulting from overlapping regions of deletion, can be identified for the patients, as a group.
Chromosomal duplications. Duplications within a chromosome represent tandemly duplicated regions of genetic material resulting in a partial trisomy and also lead to clinical phenotypes.
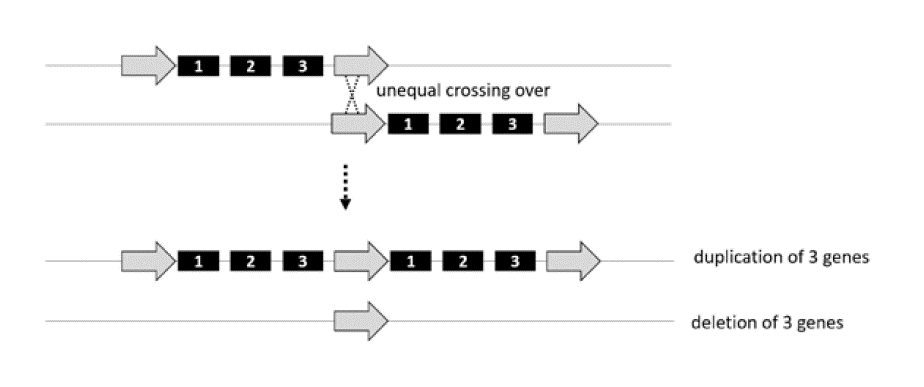 Generation of copy number variants by unequal meiotic crossing-over. Deletions and duplications of chromosome material may result from unequal crossing-over during meiosis or may occur in the offspring of reciprocal translocation carriers.
Generation of copy number variants by unequal meiotic crossing-over. Deletions and duplications of chromosome material may result from unequal crossing-over during meiosis or may occur in the offspring of reciprocal translocation carriers.
Unequal crossing-over during meiosis is a major mechanism for the de novo generation of deletions and duplications. They often occur at repetitive sequences, which are found throughout the genome and can serve as recombination hotspots during meiosis. Ordinarily, crossing-over during meiotic recombination takes place within a pair of homologous chromosomes that line up exactly, base pair-by-base pair. As the figure illustrates, however, when there is a repetitive sequence (open arrow), the two chromosomes within the pair may align out of registration. If that happens, the resulting recombination event can create a deletion or duplication. The same regions that are deleted can also be duplicated. For example, there is a 22q11.2 duplication disorder.
Copy number variant disorders therefore very often arise de novo through this mechanism. Once they are generated, then they are transmitted with autosomal dominant genetics.
SLO 4 Distinguish constitutional from acquired chromosomal abnormalities
In any discussion of cytogenetics, it is important to first distinguish between “constitutional” and “acquired” chromosomal abnormalities. Constitutional refers to the genetic make-up at birth. These abnormalities are usually present in every cell of the body, are often inherited, and are at risk of being transmitted to children. Acquired (somatic) chromosomal abnormalities usually occur after birth (or at least post-zygotically, after conception), are in a limited distribution of the cells of the body (often times just in a tumor, for example), are not inherited, and are not at risk for being transmitted to future generations.
Acquired chromosome abnormalities may occur in various types of hematologic malignancies and solid tumors.
An example of this type of phenomenon is the reciprocal t(9;22) translocation between chromosomes 9 and 22 seen in patients with chronic myelogenous leukemia (CML). This translocation alters the position of two proto-oncogenes (BCR and ABL) resulting in formation of a fusion gene product with novel catalytic activity. The derivative chromosome 22 is referred to as the “Philadelphia chromosome,” and is a marker for CML. The BCR-ABL fusion gene product creates a novel kinase that is uniquely targetable by an effective drug therapy, the first-in-class for which is imatinib (tradename, Gleevec), which has considerably improved prognosis for CML patients.
Chromothripsis. This is a recently discovered bizarre and spectacular somatic cell phenomenon, predominantly described in cancer cells, in which multiple chromosomes shatter into thousands of smaller pieces and are imperfectly reassembled, leading to an abnormal number of chromosomes and numerous segmental chromosomal translocations, deletions, and insertions throughout the genome.
SLO 5 List clinical scenarios for which a chromosomal abnormality should be considered
So, when is it appropriate to order a chromosome analysis? The following are examples of individuals who may have a chromosome abnormality or carry a rearrangement that may predispose to offspring with a chromosome abnormality:
- Individuals with multiple congenital malformations, intellectual disability, and growth failure (typical characteristics of an autosomal chromosomal imbalance).
- Confirmational testing of suspected chromosome abnormalities based upon their clinical phenotype, for example to confirm a clinical suspicion of 22q11.2 deletion syndrome or Down syndrome.
- Couples with recurrent pregnancy loss (suspecting that one parent may carry a balanced rearrangement).
- Infertility without evidence of a pregnancy loss
- Females with short stature or primary amenorrhea (possible X chromosome abnormality, i.e., Turner syndrome).
- Males with infertility (possibly Klinefelter syndrome or Y chromosome abnormality).
- Patients with ambiguous genitalia, where genetic definition of sex may be helpful in conversations related to gender choice.
- Patients with hematologic malignancies (to aid in diagnosis, prognosis, and treatment of a specific malignancy based upon the particular acquired chromosome abnormality) and certain solid tumors.
Feedback: