
2 The Pituitary Gland
Introduction
Neuroendocrinology is primarily focused on the complex interplay between the hypothalamus (nervous system) and the pituitary gland (endocrine system), the site where neural activity merges with hormonal physiology. This chapter will mostly focus on the function and abnormalities of growth hormone and prolactin.
Embryology
The pituitary gland has a dual embryological origin. Anatomy and embryology of the hypothalamus and pituitary gland will be covered in Head, Neck and Gut. The anterior portion arises from Rathke’s pouch, an evagination from the foregut in the region of the developing third ventricle within the diencephalon. These ectodermal cells form the anterior pituitary, also known as the adenohypophysis. At the same time, an evagination of the neural tissue of the embryo comes to lie over the posterior surface of the anterior pituitary, giving rise to the posterior pituitary also known as the neurohypophysis. The narrow zone between the adeno and neurohypophysis form the pars intermedia. Neuropeptides are manufactured in the hypothalamus and are secreted into the small portal circulation, known as the hypophyseal portal system, where they are carried down the pituitary stalk, to the anterior pituitary. This allows high concentrations of the hypothalamic neuropeptides to reach the pituitary gland.
Anatomy
The hypothalamus is composed of fields of neurons termed nuclei, and numerous fiber tracts entering and leaving the area. Many of the hypothalamic neurons are neurosecretory cells, with the capacity to secrete peptides and hormones. These neurons release both stimulatory and inhibitory hormones to the proximal hypothalamic-pituitary portal vessels.
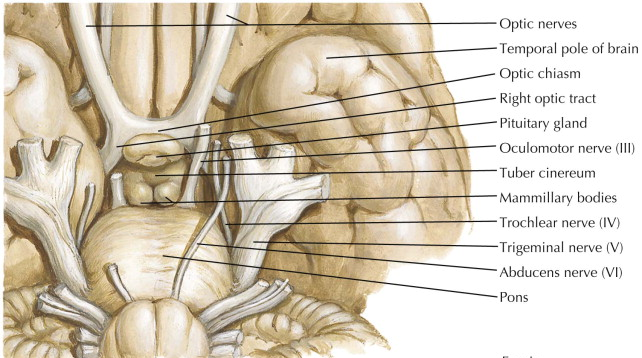
The pituitary lays on the end of the stalk (also known as the infundibulum) within the sella turcica, separated from the base of the brain by the diaphragm sellae, a fibrous membrane bridge (Figure 1). Structures near the sella turcica include the optic chiasm superiorly, the carotid sinuses laterally, the sphenoid sinus anteriorly and the third ventricle posteriorly. Tumors of the pituitary gland can exert mass effect on the surrounding structures. Thus, a basic knowledge of pituitary anatomy is necessary to fully understand pituitary pathology.
Connections between the pituitary and hypothalamus are both neural and vascular (Figure 2). The neural connection is through two neurosecretory tracts. These carry neurosecretory axons from neurons to their endings in the posterior pituitary. The vascular connection is via the primary vascular plexus and the portal hypophyseal vessels. Anterior pituitary hormones reach the peripheral circulation through venous drainage posteriorly through the cavernous sinus into the superior and inferior petrosal sinuses to the jugular vein.
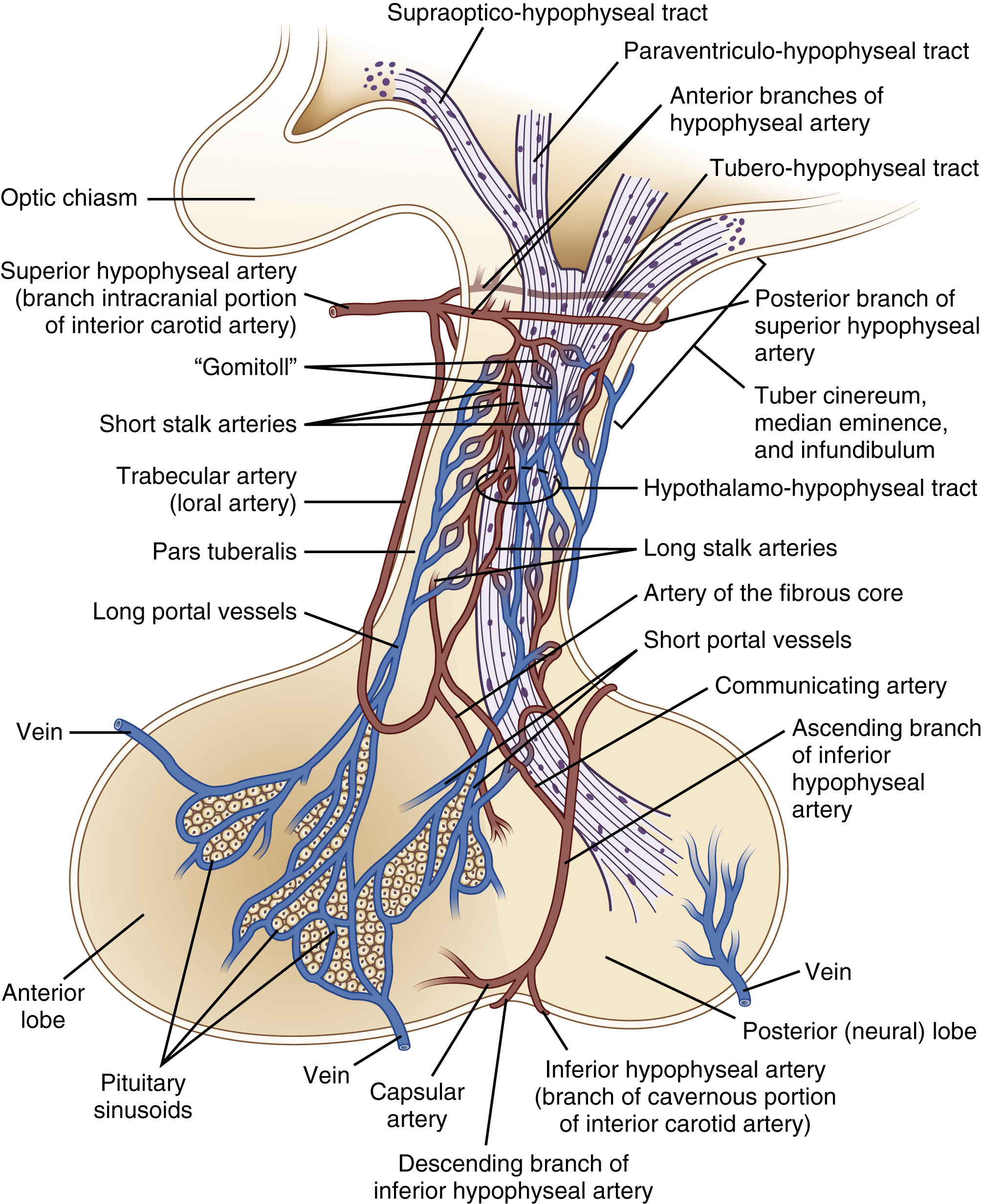
The anterior pituitary has a portal system, delivering hormones from the hypothalamus to the anterior pituitary. The posterior pituitary does not. Neurons from the posterior pituitary travel through the pituitary stalk to release neurotransmitters and other signals into the posterior pituitary.
Pathology
Pathology of the pituitary gland is covered in your Histopathology Pressbook. In brief, pituitary gland tissue is differentiated between anterior and posterior pituitary tissue. In the anterior pituitary, cells are classified by hormone secretion, their staining (though this is more historical), and the specific transcription factors (Pit1, SF1, TPit) that regulate development of these cell lineages.
Anterior pituitary
Control of anterior pituitary hormones
The hypothalamic peptides (termed “releasing hormones” [RH] or “release inhibiting hormones” [RIH]) that are released into the portal circulation to the anterior pituitary are released in extremely small amounts and are difficult to measure in the peripheral circulation. The generic notation “XRH” is used to note the hypothalamic hormones that stimulate hormones from the anterior pituitary. Examples of releasing hormones are growth hormone releasing hormone (GHRH), thyrotropin releasing hormone (TRH), and corticotrophin releasing hormone (CRH) – see table 1 below. The hypothalamic releasing hormones (RH) travel via the portal circulation to the anterior pituitary where each binds with its specific receptor displayed on the surface of its own target cell. These releasing hormones then cause the release of the specific “stimulating hormones” (aka SH) from the anterior pituitary into the systemic circulation. An example of stimulating hormones are thyroid stimulating hormone (TSH), adrenocorticotrophic hormone (ACTH). Releasing hormones typically signal via cell surface receptors, leading to activation of adenylate cyclase and generation of cAMP (the exceptions are GnRH and TRH). These neuronal circuits function with an episodic pattern, which modulates the action of the target pituitary hormone. A summary of the major hypothalamic peptide hormones is shown in Table 1.
|
Hypothalamic hormone |
Signaling pathway |
Function |
|
Thyrotropin releasing hormone (TRH) |
GPCR (Gq) Activates PLC Cleaves PIP2 Generates 2nd messenger IP3 |
Releases TSH and PRL and, to some extent, GH. |
|
Gonadotropin releasing hormone (GnRH) |
GPCR (Gq) Activates PLC Cleaves PIP2 Generates 2nd messenger IP3 |
Releases LH and FSH. |
|
Corticotrophin releasing hormone (CRH) |
GPCR (Gs) Activates AC Generates 2nd messenger cAMP |
Release ACTH (cadrenocorticotropin hormone/corticotropin), beta-endorphin, alpha-melanocyte stimulating hormone, and beta-lipotropin |
|
Somatostatin (SS) (aka growth hormone release inhibiting hormone [GHRIH]) |
GPCR (Gi) Inhibits AC Inhibits cAMP production |
Inhibits GH and TSH release. |
|
Growth hormone releasing hormone (GHRH) |
GPCR (Gs) Activates AC Generates 2nd messenger cAMP |
Releases GH. |
|
Dopamine |
Multiple receptor types |
Inhibits PRL. Dopamine is also a major neurotransmitter throughout the body. |
Hormones of the anterior pituitary
In general, the anterior pituitary hormones are much larger, more complex molecules than the hypothalamic peptides. Most pituitary hormones act as trophic hormones, which mean that they are specific for a target endocrine gland, where they stimulate the growth of the gland as well as the release of an effector hormone. Others act as direct effectors, acting on many peripheral tissues directly. They all signal via cell surface receptors. The anterior pituitary hormones are shown in Table 2. TSH, ACTH, LH and FSH are trophic hormones. These hormones cause growth in target tissues.
|
Hormone |
Signaling pathway |
Function |
|
Luteinizing hormone (LH) Follicle stimulating hormone (FSH) |
GPCR (Gs) Activates AC Generates 2nd messenger cAMP |
Stimulate ovaries and testes, release of estrogen and testosterone Stimulates growth of target glands. |
|
Thyrotropin, or thyroid stimulating hormone (TSH) |
GPCR (Gs) Activates AC Generates 2nd messenger cAMP |
Stimulates thyroid, thyroid hormone production and thyroid gland growth. |
|
Growth hormone (GH) |
Cell-surface receptor Activates non-receptor tyrosine kinase; JAK/STAT pathway Activated STAT induces gene transcription |
Stimulates growth and/or synthetic function in bone, cartilage, liver, and many soft tissues and connective tissues, IGF-1 production |
|
Prolactin (PRL) |
Cell-surface receptor Activates non-receptor tyrosine kinase; JAK/STAT pathway Activated STAT induces gene transcription |
Direct effector at breast tissue. Induces lactation. |
|
Corticotropin, or adrenocorticotropin hormone (ACTH) |
GPCR (Gs) Activates AC Generates 2nd messenger cAMP |
Stimulates adrenal cortex, production of cortisol and adrenal androgens. Stimulates growth of target glands. |
|
Melanocyte stimulating hormone (MSH) |
GPCR (Gs) Activates AC Generates 2nd messenger cAMP |
Direct effector. Stimulates melanin synthesis and other functions
|
Feedback inhibition
The hypothalamus-pituitary-endocrine organ axis is integrated to maintain very stable levels of endocrine hormones, as was discussed in the introductory chapter. The hypothalamus receives information regarding the status of the organism, the environment, and feedback data from the effector systems themselves. The endocrine system provides feedback via circulating hormones, which exert either positive or negative feedback on the system.
The physiology of the trophic hormones and their target glands will be considered in later lectures of this course. In the remainder of this chapter we will review the physiology of growth hormone and prolactin, and the diseases associated with growth hormone and prolactin excess. We will consider general pituitary pathology including pituitary dysfunction due to pituitary tumors. We will also review disorders of the posterior pituitary.
Growth hormone (GH)
The pituitary is essential for normal growth. When young animals are deprived of hormones secreted from the pituitary gland, they cease to grow. If the hormones of the target glands – thyroid, gonads, adrenal cortex – are replaced, normal growth is not restored until GH is replaced as well. However, if GH is given without replacing the other hormones, normal rates of growth cannot be achieved. Therefore, although GH is an essential factor for growth, growth also depends upon several other hormones acting in concert. If a child develops hypopituitarism (impaired pituitary function), replacing GH is essential for normal growth. Once an animal is fully-grown, GH is not essential for life. However, GH does have biological activity in adulthood. It continues to function as a counter-regulatory hormone to protect against hypoglycemia (low blood glucose). Adults who are GH-deficient lose muscle mass and strength, gain excess intra-abdominal fat, and lose bone density.
Regulation of GH secretion
Growth hormone secretion is regulated via a pituitary hypothalamic feedback loop (Figure 3). Growth hormone releasing hormone (GHRH) is released from the hypothalamus, which binds to the GHRH receptor on the somatotroph cells of the anterior pituitary. These cells release GH into the systemic circulation, where it binds to GH receptors at different sites causing release of insulin-like growth factor-1 (IGF-1), primarily from the liver. IGF-1 feeds back at the pituitary and hypothalamus to inhibit release of both GH and GHRH via a negative feedback loop.
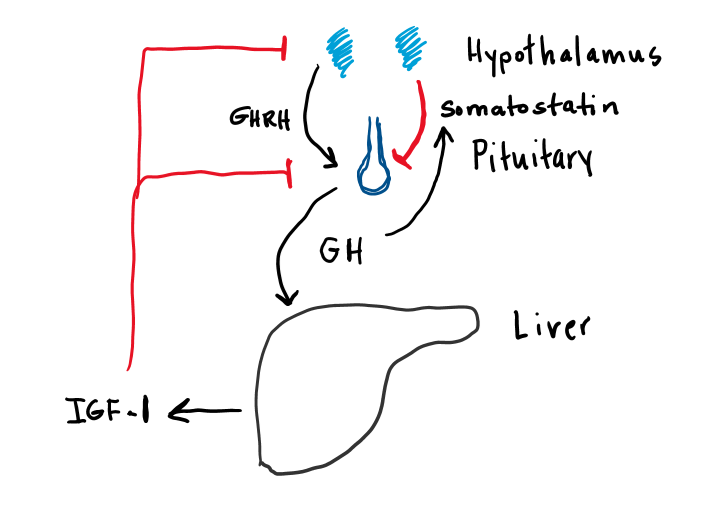
There are at least two other important regulators of GH secretion. Somatostatin is an inhibitory hormone produced in the hypothalamus. Its secretion is stimulated by GH and IGF-1 and inhibits GH secretion via a negative feedback loop. Ghrelin receptors are also present on somatotroph cells of the pituitary. Ghrelin, which is produced in the stomach, stimulates GH secretion, suggesting possible nutritional regulation of GH secretion. As with most of the anterior pituitary hormones, GH is secreted in periodic pulses. The basic pattern of secretory pulses can be modulated by a number of factors, as detailed in Table 3.
|
Factors that increase GH secretion |
Factors that inhibit GH secretion |
|
Sleep |
Beta 2-agonists |
|
Exercise |
Alpha 2-antagonists |
|
Stress |
Hyperglycemia (high blood glucose) |
|
Arginine |
|
|
Beta blockers |
|
|
Hypoglycemia (low blood glucose) |
|
Actions of Growth Hormone
Insulin-like growth factor 1 (IGF-1) is the main effector of GH. GH stimulates the synthesis and release of IGF-I by the liver. IGF-I produced in response to GH circulates in association with a group of binding proteins. Caloric deprivation and insulin deficiency reduce IGF-I production.
IGF-1 binds to its receptors on target tissues. The IGF-1 receptor is a receptor tyrosine kinase. Binding of IGF-1 leads to dimerization of the receptor and cross-phosphorylation of tyrosine residues, which leads to the downstream effects of IGF-1. The major action of IGF-I is to stimulate growth.
The growth promoting effect is particularly strong for bone but in the presence of excess GH (and IGF-I), virtually all soft tissues grow at a more rapid rate than normal. GH also has several direct metabolic effects, all of which are catabolic (with the purpose of releasing energy). Growth hormone increases lipolysis, decreases triglyceride uptake, increases gluconeogenesis and increases glycogenolysis. Growth hormone excess can cause insulin resistance and high blood glucose, both contributing the development of diabetes mellitus.
Growth hormone and IGF-1 have antagonistic metabolic effects. IGF-1 has anabolic effects and leads to fuel storage. IGF-1 decreases lipolysis, increases lipogenesis, increases glycogen synthesis and increases GLUT4 translocation in the plasma membrane.
Diseases of Growth Hormone Secretion
Acromegaly and Gigantism
When a GH-producing tumor of the pituitary develops and functions autonomously, accelerated growth of bones and soft tissues takes place. If the adenoma (benign pituitary tumor) occurs during childhood, the condition called “gigantism” develops. If untreated, the individual may gain great height (Figure 4a). When GH excess occurs in childhood, the long bones are able to reach such enormous lengths because growth is occurring before closure of the epiphyseal plates, a process triggered by the increased gonadal steroids present in puberty. GH-producing adenomas appearing after puberty (and closure of the growth plates) give rise to the disease “acromegaly.” Acromegaly causes a distorted pattern of bony overgrowth with widening and thickening instead of simple elongation of the long bones (Figure 4b). The annual incidence of acromegaly in the US is 4.6 cases per 1 million people per year.
With an overproduction of growth hormone, the changes in appearance may occur slowly over a long period of time, so that the person and their friends and relatives might not notice. Overgrowth of the mandible can cause wide gaps between the teeth and an “underbite,” so that the diagnosis may occasionally be made by an astute dentist. The patient may notice that they require increased size of rings, shoes, or hats, which had been stable through earlier adult life. Other bones such as the mandible and the membranous bones of the skull grow in all dimensions. Because of the bony distortion around weight-bearing joints and in the spine, a painful degenerative arthritis is a prominent clinical feature. The soft tissues also grow and become distorted in appearance. The earliest changes are coarsening of the facial features and widening of the feet and hands. Sweating is excessive, sebaceous activity increases, and lips and tongue become enlarged.
Both skeletal and cardiac muscle are stimulated to grow, but the hypertrophy of muscles is accompanied by a myopathy (muscle damage), causing weakness and fatigue. Left and right ventricular hypertrophy (enlargement of the right and left pumping chambers of the heart) can lead to heart failure and is the main cause of premature death in those not appropriately treated. Sodium retention manifests as hypertension (high blood pressure) in 35% of patients with acromegaly, and the insulin resistant properties of GH as diabetes mellitus in 20%. Because of the non-specific nature of many of the symptoms and the slow evolution over time, the time between onset of symptoms and diagnosis is on average 9 years for adults with growth hormone excess.


Growth Hormone Deficiency
Deficient secretion or activity of GH will lead to growth failure in children. This is generally diagnosed after growth arrest is noted on pediatric growth charts. Replacement of GH in childhood is critical for normal growth (Figure 5).
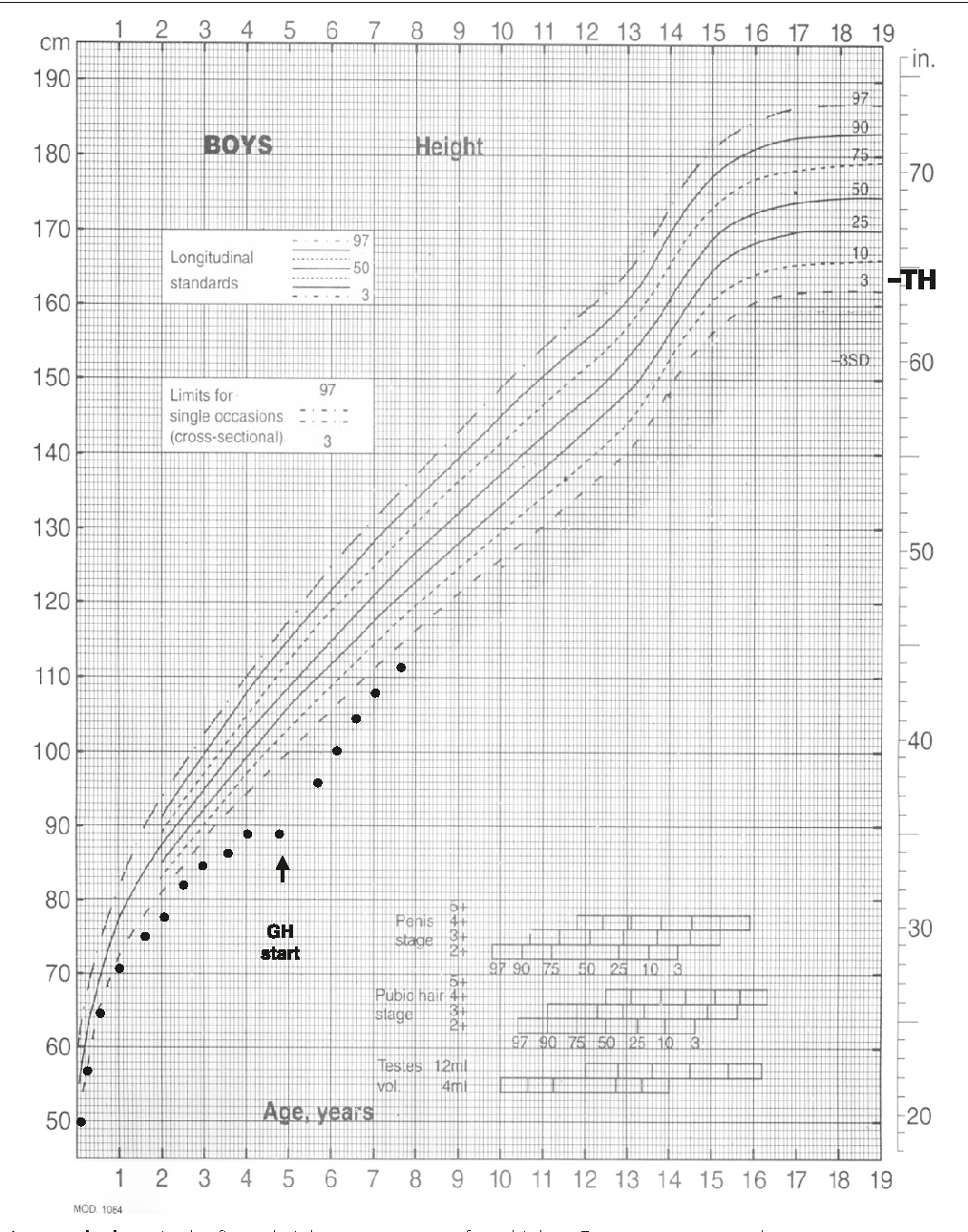
Acquired adult GH deficiency is usually a consequence of pituitary dysfunction from injury, surgical manipulation, or an expanding sellar mass. Since linear growth is already completed in adulthood, GH deficiency in adults results in several metabolic irregularities (hyperlipidemia, decreased lean muscle mass, decreased bone mineral content). Replacement of GH in adults with GH deficiency is controversial due to unclear clinical benefits and many side effects. GH therapy improves fat free mass, bone density and general well-being in adults with growth hormone deficiency.
Diagnosis of Growth Hormone Disorders
As previously mentioned, GH levels can fluctuate throughout the day, so measuring random levels is not helpful in diagnosing GH disorders. Because plasma levels of IGF-1 are more stable than GH itself, the measurement of plasma IGF-I is a useful screening test for diagnosing GH abnormalities. IGF-1 would be high in conditions of GH excess and low in GH deficiency.
If IGF-1 levels (the initial screening test) are abnormal, confirmatory tests are needed. One of the central tenets of endocrinology when considering the diagnosis of abnormal hormone levels is that if you suspect a hormone level is high, try to suppress it. If you suspect it is low, try to stimulate it. If hormone levels are inappropriate due to underlying pathology, they will not respond normally to stimulation or suppression. Glucose is a potent regulator of GH secretion, with low levels stimulating GH secretion and high levels suppressing it. Thus, if you suspect GH excess (with an elevated IGF-1 on screening tests), you would attempt to suppress GH secretion by inducing high blood glucose (hyperglycemia). If GH fails to suppress with hyperglycemia, this confirms the diagnosis of GH excess and likely pituitary adenoma. This is illustrated in figure 6a.
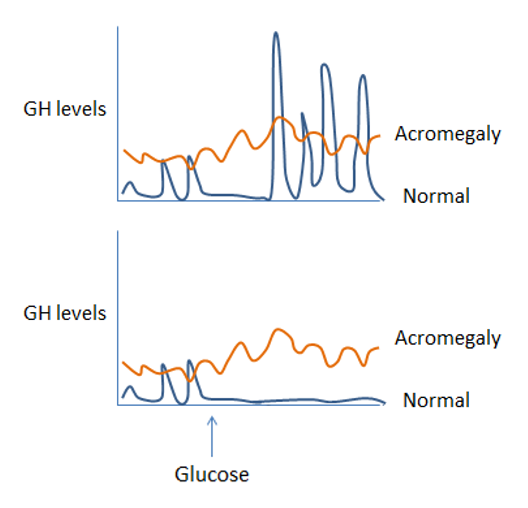
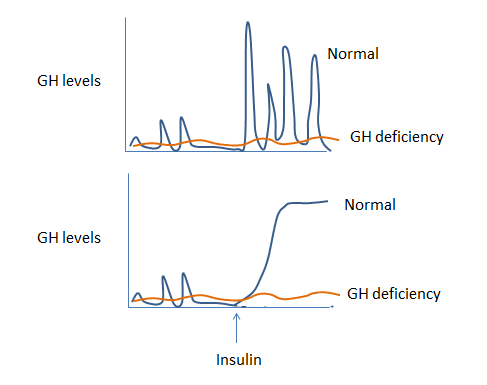
Similarly, if you suspect GH deficiency (with low IGF-1 on your screening test), you may try to stimulate GH secretion by inducing hypoglycemia (low blood glucose) with insulin in an “insulin tolerance test”. Failure to increase GH with hypoglycemia confirms GH deficiency (figure 6b).
Treatment of Growth Hormone Excess
An understanding of the anatomy, physiology and biochemistry of growth hormone has facilitated the development of a variety of approaches to the treatment of growth hormone excess or deficiency (Figure 7).
Treatment of acromegaly is necessary to reduce the morbidity and mortality associated with excess GH secretion. Patients with acromegaly are prone to diabetes, hypertension, osteoarthritis, cancer and premature death from heart failure. Normalization of excess GH reduces mortality and severity of the metabolic, cardiovascular, and orthopedic consequences of GH excess.
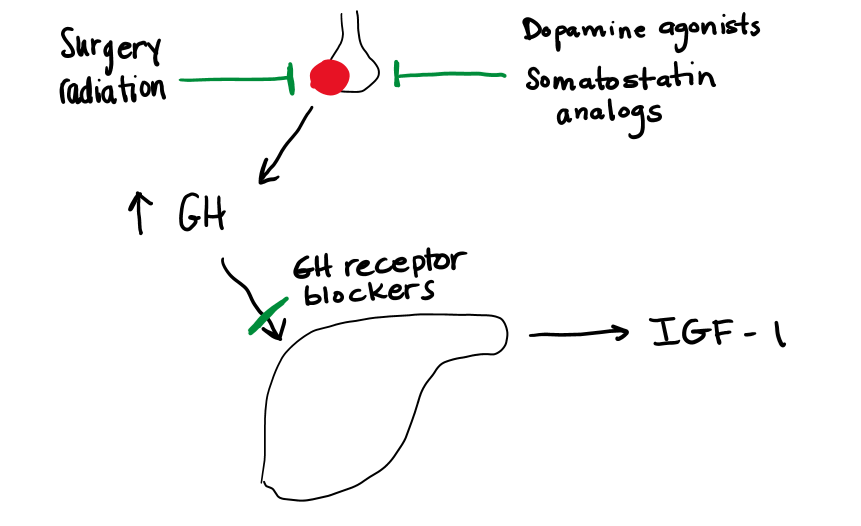
Surgery Removal of the growth hormone secreting pituitary tumor by transsphenoidal pituitary surgery is the first line option for treatment. In the best hands, surgical cure rates can reach 70-80%. For larger tumors (macroadenomas) with suprasellar and lateral extension, cure rates decrease. Pituitary injury and subsequent hypopituitarism are higher in this setting. Successful cure is based on surgeon expertise, tumor size and location.
Radiotherapy can target a tumor with a margin of error between 3-5 mm, depending on the type of radiation used. However, for large tumors and tumors that encroach within 3-5 mm of the optic chiasm, there is a risk of optic nerve damage which limits use of radiation. All forms of radiotherapy are slow and take time to normalize GH excess. The risk of hypopituitarism is almost 100% over time due to radiation damage to the normal pituitary cells.
Dopamine agonists (selective D2 receptor agonists, such as bromocriptine and cabergoline) are very effective in the treatment of prolactin-secreting tumors (see below) and may have some effect in inhibiting GH secretion in acromegaly. They have the advantage of being oral drugs, but are much less effective than the somatostatin analogs. Dopamine can inhibit GH secretion, but is not used therapeutically. See figure 11, below.
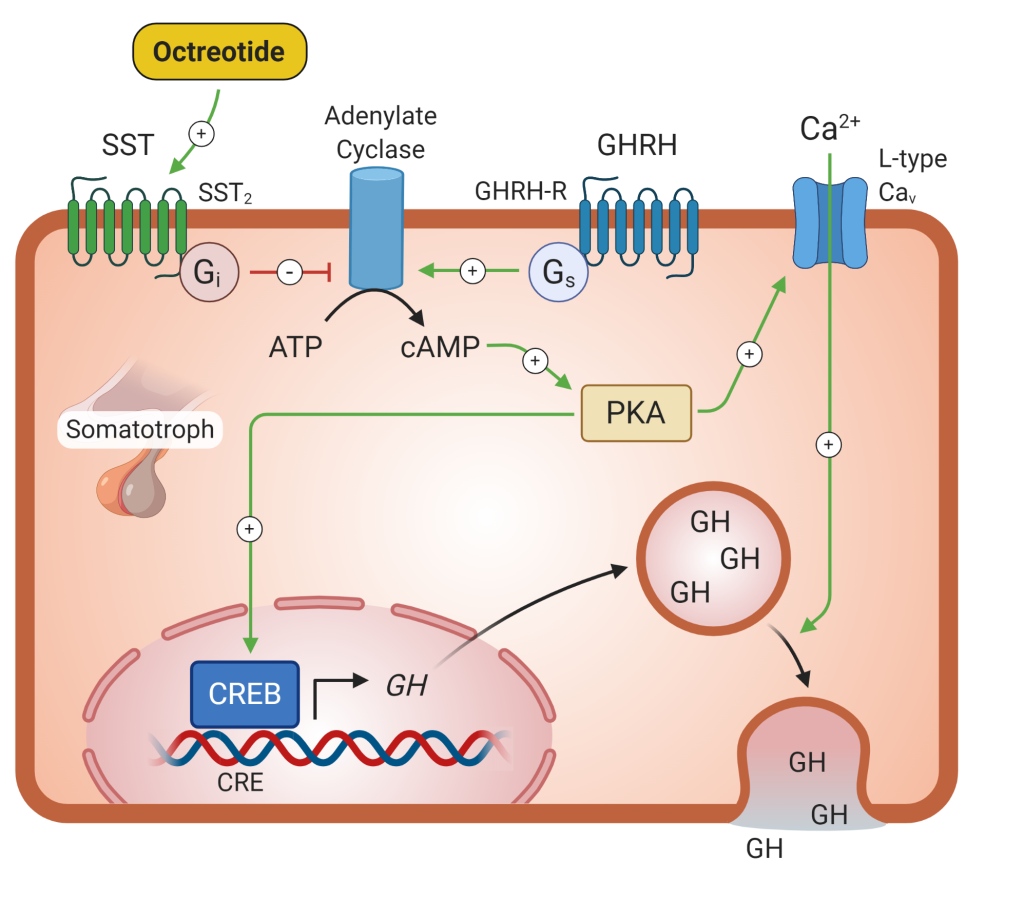
Somatostatin analogs (such as octreotide, lanreotide, and pasireotide) (Figure 8) are very effective in reducing GH secretion in acromegaly. Somatostatin is a direct inhibitor of GH secretion and most of these tumors express somatostatin receptors. Most of these medications need to be administered by injection. These agents will also have an impact on tumor growth, causing a decrease in tumor size or arresting growth in nearly 85% of tumors. Tumor regression is usually modest, at best. Common side effects with the somatostatin analogs include transient gastrointestinal symptoms, which resolve within the first few weeks of therapy, increased risk of gallstones, and hyperglycemia due to inhibition of insulin release.
Growth hormone receptor antagonists (pegvisomant, Figure 9) block the effect of GH and lead to decreased IGF-1 levels. Because this agent acts via GH receptor inhibition, GH levels rise in response to falling IGF-1. In this case, GH levels cannot be followed to assess cure. Rarely (2% of cases) this agent may cause tumor growth. Pegvisomant may cause elevated liver enzymes, which will resolve on discontinuation of the medication. It is a once daily subcutaneous injection, and some patients do experience injection site reactions.
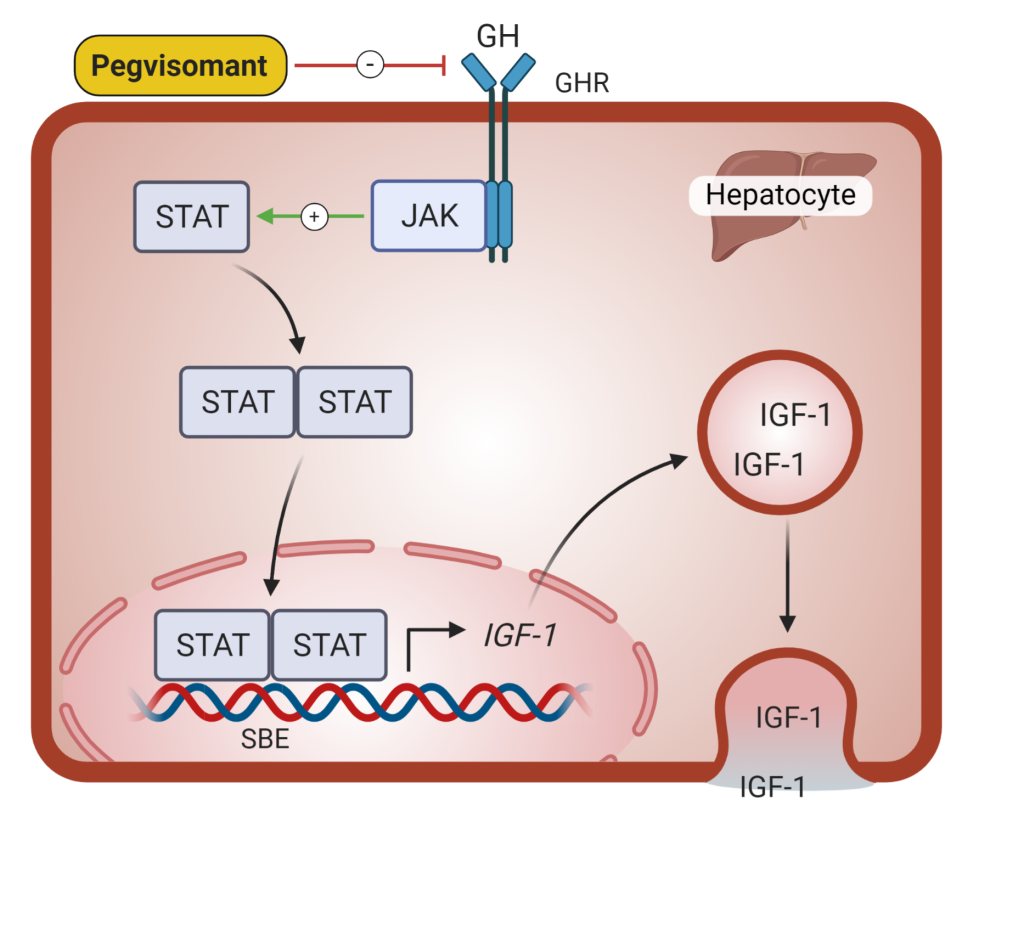
Treatment of growth hormone deficiency
Recombinant human growth hormone (rhGH) can be manufactured synthetically and given as a subcutaneous injection (typically daily but there are some longer acting versions available that can be injected weekly). This is a necessary treatment for childhood GH deficiency. As discussed earlier, adults with with growth hormone deficiency may benefit from growth hormone replacement but the positive impacts are not as clear as they are in children. More controversial is the treatment of normal children who are of short stature or older adults who have a natural decline in their GH. There are no significant side effects with rhGH when used at typical treatment doses. When doses are given in excess, side effects will be similar to the clinical syndrome seen in acromegaly. Off label use of rhGH may be used by body builders and athletes with the intention of increasing muscle mass. The off-label use of rhGH for this purpose is dangerous and ill advised.
Prolactin (PRL)
Prolactin is a protein secreted from the lactotroph cells of the anterior pituitary. The normal serum level of prolactin varies by laboratory but usually ranges between 15-20 ng/ml in genetic females and 10-15 ng/ml in genetic males. Prolactin is necessary for breast development to promote lactation during nursing. Serum levels are significantly elevated during pregnancy due to the stimulatory effects of estrogen on the lactotroph cells. Elevated serum levels can lead to an altered gonadotrophin releasing hormone (GnRH) pulse frequency and cause hypogonadotropic hypogonadism.
Regulation of prolactin secretion
Prolactin secretion, similar to other anterior pituitary hormones, is regulated by the hypothalamus. Unlike the other anterior pituitary hormones however, prolactin secretion is under tonic inhibitory control (figure 10). The neurotransmitter dopamine secreted from the hypothalamus inhibits prolactin release from the pituitary. Loss of dopamine leads to an increase in prolactin levels. Prolactin levels can be greatly affected by D2 dopamine receptor agonists and antagonists. Thyrotropin releasing hormone (TRH) can also stimulate prolactin release from the lactotrophs, so prolactin may be elevated in primary hypothyroidism because the lack of negative feedback from the thyroid gland causes elevation of TSH and TRH. This is typically only seen in severe cases of primary hypothyroidism. Estrogen has a direct stimulatory effect on lactotroph growth and secretion and thus increases prolactin production. Suckling of the breast stimulates prolactin secretion via a neural afferent pathway.
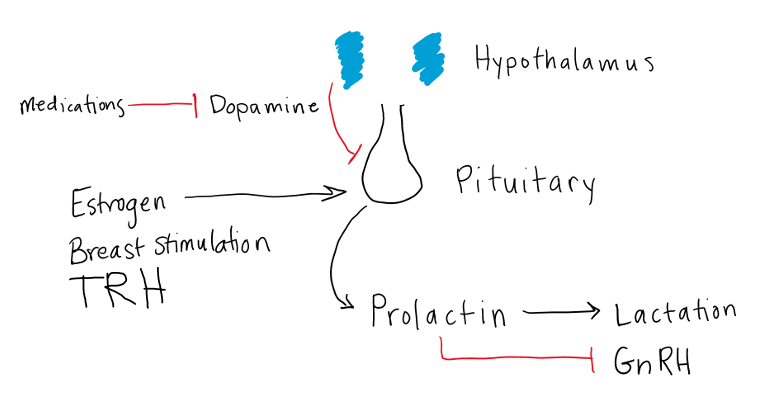
Disorders of Prolactin Secretion
Prolactin deficiency
A decrease in serum prolactin may be a consequence of certain causes of hypopituitarism. Infiltrative (sarcoidosis, fungal infection) processes or destruction of pituitary tissue with infarction or hemorrhage can damage the lactotrophs, leading to decreased prolactin, as well as low levels of other pituitary hormones. The only known consequence of prolactin deficiency is a lack of milk let-down in the postpartum period. This clinical manifestation is a hallmark of pituitary vascular infarction, known as Sheehan’s syndrome, following obstetrical hemorrhage (see below).
Prolactin excess (hyperprolactinemia)
There are multiple causes of elevated prolactin, which are discussed in more detail in the next section. Hyperprolactinemia causes hypogonadotropic hypogonadism (low estrogen or low testosterone) by decreasing GnRH which leads to decrease in LH and FSH. In genetic females, low estrogen from hypogonadotropic hypogonadism can present as oligomenorrhea (irregular periods) or amenorrhea (lack of periods) and decreased fertility. The prolactin elevation can cause galactorrhea (inappropriate production of breast milk). In genetic males, prolactin elevation will also cause suppression of testosterone, with a resultant decrease in libido and sometimes erectile dysfunction. Hyperprolactinemia often decreases fertility due to decreased sperm production.
Causes of Hyperprolactinemia
Hyperprolactinemia may be a consequence of one or multiple of these conditions (1) loss of dopamine inhibition, (2) increased prolactin production (physiologic or pathologic), or (3) decreased prolactin clearance. Most causes fall in categories (1) or (2).
Physiologic hyperprolactinemia
Pregnancy results in elevated estrogen levels that stimulate lactotroph growth and increased prolactin secretion. Prolactin is renally cleared, so renal failure can lead to hyperprolactinemia via diminished clearance of the hormone. Primary hypothyroidism induces an increase in TRH that in turn has a stimulatory effect on lactotrophs and can cause elevated prolactin. Chest wall injuries and nipple manipulation create stimulatory afferent signals similar to suckling. With the exception of pregnancy, where levels can reach very high values, levels of prolactin rarely exceed 50-60 ng/ml from physiologic causes.
Diminished dopaminergic inhibition of lactotrophs
Inhibition of the inhibitory effects of dopamine on the lactotrophs will lead to an increase in prolactin levels. Injury to hypothalamic neurons from tumor or other causes can lead to diminished dopamine production. Use of pharmacologic agents which lead to depletion of central dopamine stores (reserpine, methyldopa) or which act as D2 dopamine receptor antagonists (phenothiazines, butyrophenones, metoclopramide, domperidone) can lead to hyperprolactinemia. There are many medications that decrease dopamine and subsequently increase prolactin. Any insult that reduces portal blood flow (“stalk compression”) will block dopamine access to lactotrophs and result in prolactin elevation. Common causes of stalk compression are trauma, sellar and suprasellar tumors, metastatic tumors, and inflammatory processes. When stalk compression is the cause of dopamine elevation this is known as “stalk effect”. Diminished dopaminergic inhibition usually results in prolactin levels between 30-100 ng/mL.
Lactotroph cell tumors or prolactinomas
Prolactinomas are the most common type of functional pituitary tumor, comprising 25-40% of all functional pituitary adenomas. They are further classified by size, with microprolactinomas measuring less than 1 cm in maximal diameter and macroprolactinomas greater than or equal to 1 cm. Prolactin levels are directly proportional to tumor size, with microprolactinomas having prolactin levels generally less than 200 ng/dl, and macroprolactinomas manifesting serum prolactin levels from 200 ng/ml to levels in the thousands. Macroprolactinomas may also present with compressive symptoms due to tumor expansion from the sella, such as visual loss or cranial nerve defects.
A detailed history and physical including a medication profile can usually differentiate among the different causes of prolactin excess. The serum prolactin level is an important clue to distinguish between the different causes of hyperprolactinemia, particularly in determining whether a pituitary adenoma is a prolactin-secreting adenoma or non-secreting adenoma. This is crucial in deciding on appropriate treatment, as discussed in the next section.
Treatment of hyperprolactinemia
Treatment of hyperprolactinemia is dependent on the cause. The reasons to treat hyperprolactinemia fall into three categories: 1) to reduce mass effect if the cause of hyperprolactinemia is a macroprolactinoma, 2) to reverse hypogonadism (low estrogen or low testosterone) and 3) to diminish galactorrhea. Unlike other pituitary adenomas, prolactinomas do not require surgical treatment. Prolactinomas are amenable to medical management with dopamine agonists. You will recall from Figure 10, that dopamine secreted from the hypothalamus decreases prolactin secretion. By increasing dopamine with dopamine agonist medications (Figure 11), such as cabergoline and bromocriptine, prolactin will decrease. With few exceptions, all patients requiring therapy for prolactinomas should be offered medical therapy; only if this therapy fails or is not tolerated should surgical resection of a prolactin-secreting tumor be considered. Side effects (dizziness, headache, nausea) are less common with cabergoline, so this is the preferred medication when treating prolactinomas.
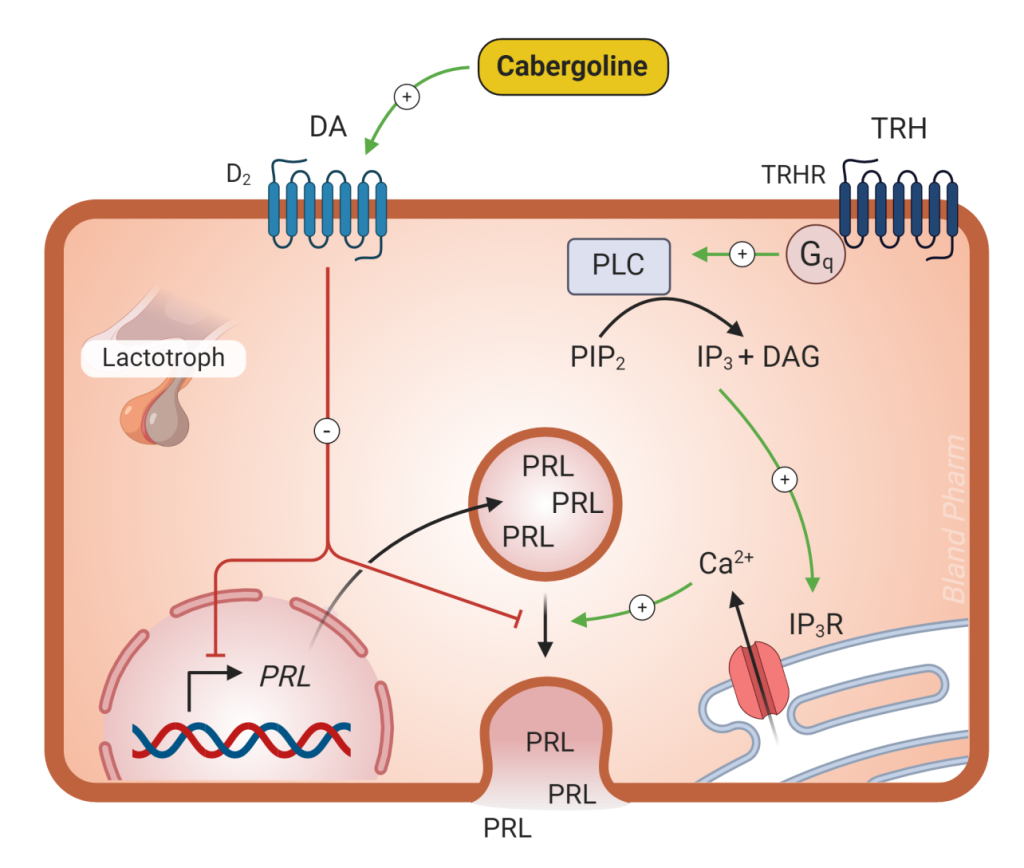
When the cause of prolactin elevation is not due to a prolactinoma, the underlying cause should be addressed. In the case of a pituitary tumor that is causing stalk effect, surgical resection may be considered. For medication-induced prolactin elevation, a dose change or medication change may be considered if there are clinical effects from the prolactin elevation. Dopamine agonist medications will work to decrease prolactin elevation no matter the cause but they will not lead to pituitary tumor shrinkage in most cases of prolactin elevation that are not due to a prolactinoma.
Hypopituitarism
Causes
An underproduction of any or all of the hormones of the anterior and posterior pituitary can lead to a variety of clinical presentations. In complete hypopituitarism, patients present with symptoms of hypothyroidism (low thyroid), hypoadrenalism (low cortisol), growth failure and hypogonadism. These will be discussed in more detail in the chapters pertaining to the specific hypothalamic-pituitary-endocrine organ axis. Common causes of hypopituitarism are listed in Table 4. It is important to realize that any of these conditions can lead to partial (some hormone deficiencies) or complete hypopituitarism (deficiency of all pituitary hormones – both anterior and posterior). Pituitary apoplexy will be described in the following section. It is important to understand the difference between hypopituitarism caused by pituitary apoplexy and a pituitary adenoma.
|
Causes of Hypopituitarism |
|
Pituitary tumors |
|
Pituitary apoplexy |
|
Craniopharyngioma |
|
Trauma |
|
Surgery |
|
Radiotherapy |
|
Infiltration (eg. Sarcoidosis) |
|
Infection (eg. Tuberculosis) |
|
Infarctions (eg. Sheehan’s) |
|
Developmental defects |
Diagnosis
Clinically, hypopituitarism presets with either symptoms of the individual hormone deficiencies (which will be covered in detail in the chapter on the individual hormones), symptoms of the underlying cause (mass effect with pituitary tumors, for instance) or in children, as growth arrest, failure to thrive or delayed puberty.
There are two methods for diagnosing hypopituitarism. For those hormones that have stable serum levels, you can obtain a concomitant random sampling of the pituitary hormone (LH, FSH, TSH) and effector gland hormones (estrogen, testosterone, thyroid hormones). Measuring the pituitary hormone alone is not always helpful, as this can sometimes be “inappropriately normal” in the setting of hypopituitarism. As hormone levels drop, negative feedback decreases at the level of the pituitary. If the pituitary gland were responding appropriately to declining hormone levels secreted from the endocrine gland, there should be an increase in the pituitary hormone, in an attempt to increase stimulation of the endocrine gland. If the pituitary is not working properly, it will not be able to increase hormone production adequately. Levels may be in the normal range, but not appropriate for the low levels of effector hormones.
For those hormones with pulsatile secretion (GH, ACTH), random serum tests are not helpful in diagnosing deficiency. To diagnose deficiency of these hormones, a stimulatory test such as the insulin tolerance test for diagnosis of GH deficiency, is used and is considered the “gold standard” to test these pituitary axes. If the pituitary hormone does not increase appropriately after the administration of a substance that would typically stimulate its production, hypopituitarism should be suspected. Test for cortisol and ACTH deficiency will be discussed in the adrenal chapter.
Although any combination of hormone deficiencies can occur in hypopituitarism, there is often a characteristic progression where GH is lost first, followed by LH, FSH, and TSH. ACTH secretion is usually maintained with the greatest durability. This order of loss can be remembered with the mnemonic: Go Find The Adenoma (GH, FSH/LH, TSH, ACTH). Diabetes insipidus is usually a result of hypothalamic injury as the hypothalamus is the site of vasopressin (aka antidiuretic hormone, ADH) synthesis. The posterior pituitary is only the site of vasopressin release (see below) and not synthesis. Thus, destruction of the posterior pituitary therefore will not always lead to vasopressin deficiency.
Pituitary Tumors
Pathologic classification of endocrine tumors
Pituitary tumors are classified according to size, with those < 1 cm termed microadenomas and those > 1 cm termed macroadenomas. They are also classified by their hormone production and pituitary cell linage. A prolactin secreting tumor measuring 1.2 cm would be classified as a macro-prolactinoma.
There are three different cell lineages (defined by transcription factors) that help define pituitary adenomas (tumors). The Pit-1 transcription factor is the cell lineage for growth hormone secreting cells (somatotrophs), prolactin secreting cells (lactotrophs), and TSH secreting cells (thyrotrophs). The TPit transcription factor is the cell lineage for ACTH secreting cells (corticotrophs), and the SF1 transcription factor is the cell lineage for gonadotroph adenomas. These transcription factors are present in normal pituitary cells and are one of the characteristics used to classify pituitary adenomas (Table 5).
Pituitary adenomas occur due to sporadic mutations in the pituitary cell that leads to unchecked growth. The vast majority of cases are sporadic but some are syndromic/hereditary. A syndrome that leads to development of pituitary adenomas is MEN1 (loss of menin) which you will learn about later in this block.
| Transcription Factor
Lineage |
Normal pituitary cells | Hormones Produced | Tumor Types |
| Pit-1 lineage | Somatotrophs, lactotrophs, thyrotrophs | Growth hormone
Prolactin TSH |
•GH-secreting adenoma (acromegaly)
•Prolactinoma
•TSH-secreting adenoma (secondary hyperthyroidism)
•Mixed tumor (prolactin + GH)
|
| T-Pit lineage | corticotrophs | ACTH |
•ACTH-secreting adenoma (Cushing’s syndrome)
|
| SF-1 lineage | gonadotrophs | FSH, LH |
•Gonadotroph adenoma (usually not clinically apparent, aka “silent”)
|
Pituitary neoplasms, rare in childhood and adolescence, make up approximately 10% of all intracranial tumors. Most tumors arise from the endocrine cells of the adenohypophysis and are designated as pituitary adenomas. Pituitary adenomas are relatively common, with radiologic and autopsy studies revealing a prevalence of 10% in the general population. Many of these tumors have no clinical consequence. Pituitary adenomas may become clinically evident because of the increased secretion of a specific hormone by the tumor cells, by signs of hypopituitarism secondary to compressive destruction of the normal gland, and/or by symptoms or signs resulting from the compression of adjacent structures. Suprasellar extension, present in 10-20% of the cases, results in visual symptoms because of compression of the optic chiasm (which sits just above the pituitary gland). Extraocular tumor palsies (due to compression of the facial nerves that travel through the cavernous sinus: III, IV, V1, V2, VI) occur in 5-10% of the patients. In 5-10% of all pituitary tumors, actual invasion of neighboring structures is encountered, such as the anterior, middle, or posterior fossa, cavernous sinus, optic nerves, chiasm, nasopharynx, or nasal cavity. We prefer to designate these tumors as “invasive adenomas” rather than as carcinomas. Pituitary carcinoma is a designation limited to tumors with metastasis distant from the sella. Fortunately, carcinomas are rare.
“Pituitary apoplexy” is a rare complication of pituitary adenomas. It represents a massive hemorrhagic infarct (a localized area of dead tissue resulting from failure of blood supply) within the tumor and is most often seen in macroadenomas. This can lead to rapid decrease in all pituitary hormones and acute onset of hypopituitarism. Pituitary apoplexy is a clinical diagnosis with the constellation of a hemorrhagic sellar mass, pituitary dysfunction and cranial nerve defects due to tumor compression. The presentation is different from the more gradual onset of pituitary hormone loss from a large pituitary adenoma (or other pituitary tumor), see Table 6.
| Pituitary Apoplexy | Pituitary Adenoma | |
| Pathogenesis | Sudden infarct of known pituitary adenoma with bleeding into surrounding areas with rapid destruction of pituitary gland | Slow growing mass causes gradual loss of anterior pituitary hormones |
| Onset | Sudden | Gradual |
| Vision impact | Often impacted | Sometimes impacted |
| Anterior pituitary hormone impact | Sudden loss of all anterior pituitary hormones | Gradual loss (Go Find The Adenoma – GH, FSH/LH, TSH, ACTH) |
| Prolactin impact | Low due to complete destruction of gland | Elevated from stalk effect |
| Severity | Medical emergency | Requires treatment but not usually a medical emergency |
| Impact on ADH | Diabetes insipidus more common | Rare to have diabetes insipidus |
Clinical Manifestations of Pituitary Tumors
In any pituitary tumor, the clinical features are determined by three factors: local tumor growth, destruction of normal pituitary tissue (causing low hormone production), and production of excessive amounts of hormone (ACTH leading to cortisol excess, growth hormone leading to acromegaly or gigantism, prolactin, TSH leading to secondary hyperthyroidism, etc).
Local tumor growth – The growth of a tumor within a small bony cavity can cause pressure on important nearby structures. If the tumor extends outside the pituitary fossa it may put pressure on the optic chiasm which lies directly above (see Figure 1) the pituitary gland. Since the fibers of the optic chiasm are crossing over, coming from the medial parts of both retinae, it is the outer parts of the visual fields which are lost first (bitemporal hemianopsia, figure 12).
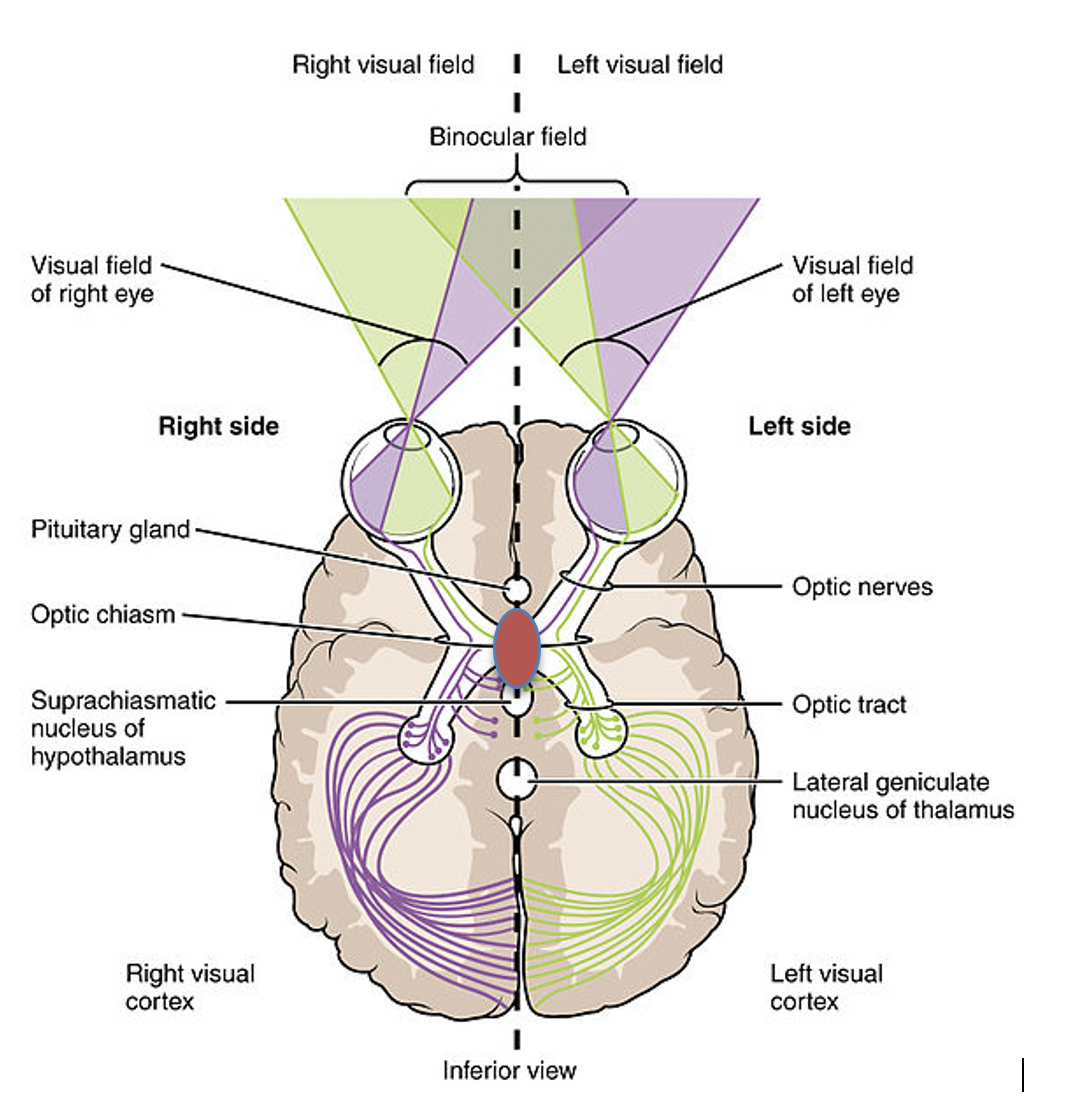
If the suprasellar extension of the tumor is asymmetrical, then the visual field defect may be more noticeable on one side than the other. Often, the patient is completely unaware that there is a visual problem until very late in the disease process, but the defect can be picked up by formal testing of the visual fields.
If the tumor invades inferiorly through the sphenoid sinus, it is possible that cerebrospinal fluid (CSF) may leak out into the nasal cavity causing CSF rhinorrhea, and raising the possibility that bacteria may gain access, causing meningitis. If a tumor extends into the cavernous sinus, palsy of the cranial nerves (III, IV, V1-2, VI) traversing the sinus may be seen. If the tumor compresses the pituitary stalk, dopamine inhibition of prolactin is lost, resulting in hyperprolactinemia (this is called “stalk effect”). The degree of local tumor growth can be detected with best resolution through MRI scanning.
Destruction of normal pituitary tissue: This causes low pituitary hormone function or hypopituitarism. As the tumor grows, the hormones affected occur in a predicted order: 1) growth hormone, 2) FSH/LH, 3) TSH, 4) ACTH. The mnemonic used to define this is Go Find The Adenoma.
Pituitary Apoplexy: Rarely, when the pituitary adenoma outgrows its blood supply, it can infarct and bleed into the pituitary gland causing a rapid and sudden loss of pituitary hormone function. All of the hormones (including prolactin) will be low (table 6). Symptoms of pituitary apoplexy include headache and vision loss (sometimes, not always).
Treatment of pituitary tumors
Specific treatment options have been mentioned earlier in the chapter for prolactinomas and growth hormone secreting adenomas. For small non-functioning pituitary tumors that do not extend within 3-5 mm of the optic chiasm or impact pituitary function, a watchful waiting approach can be employed. For tumors secreting hormone (GH,ACTH,TSH) surgical resection is the preferred option, with pharmacologic and radiation therapy reserved for surgical failure or tumor recurrence. Transsphenoidal resection offers a quick and possibly complete cure but includes some risk. Surgical resection of non-functioning pituitary adenomas is recommended if (1) the tumor is large and compressing nearby structures, (2) is growing on serial imaging studies, or (3) is causing the compromise of normal pituitary function. Treatment with gamma knife radiation is less traumatic and should stop tumor growth right away, but will take several years to achieve cure in a secreting tumor. The risk of hypopituitarism is almost 100% following radiotherapy (including gamma knife) but may take up to ten years to develop, warranting long-term monitoring of pituitary function.
If patients do develop hypopituitarism because of their pituitary adenoma, surgery or radiation, they need replacement of the missing hormones. We do not typically replace the pituitary hormone (GH is the exception to this) but instead give the effector hormone, as this tends to have a longer half-life and is more stable.
For ACTH deficiency, the patient is given cortisol replacement with hydrocortisone (or one of the other glucocorticoids) which can be taken orally, usually in two doses per day with a larger dose in the morning to mimic the diurnal variation of ACTH and cortisol secretion. Adrenal steroid replacement should take precedence over all other pituitary hormones and will be discussed in more detail in the adrenal chapter.
For TSH deficiency the patient is given synthetic thyroid hormone called levothyroxine, which can be taken orally, once a day. This will be discussed more in the Thyroid session in treatment of hypothyroidism.
As discussed above, growth hormone deficiency may or may not require treatment with rhGH.
Depending on the gender identity of the patient, LH and FSH deficiency can be treated with estrogen/progesterone or testosterone.
Diabetes insipidus, which is discussed below, is caused by deficiency of anti-diuretic hormone (ADH) action. Replacement of ADH with a medication called desmopressin via the intranasal, oral, or subcutaneous routes will relieve symptoms.
Neurohypophysis (aka Posterior Pituitary)
Synthesis and secretion of antidiuretic hormone
The brain produces a hormone, anti-diuretic hormone (ADH), to reduce renal water excretion and help regulate plasma osmolality. ADH, also called vasopressin (arginine vasopressin or AVP), is synthesized in the hypothalamus and transported within neurosecretory granules in the axons of the hypothalamic neurons to nerve endings in the neurohypophysis (posterior pituitary). The regulation and action of ADH will be covered here and in more detail in your nephrology course.
Diabetes insipidus (DI)
When there is a deficiency/decrease in ADH secretion or a problem with its action, diabetes insipidus (DI) may develop. Diabetes insipidus refers to the passage of copious amounts (> 2.5-3.5 L/day) of dilute urine without glucosuria (in contrast to diabetes mellitus which means “sweet urine”), as the absence of ADH prevents appropriate concentration of the urine. Symptoms of diabetes insipidus include nocturia and polyuria and polydipsia (night urination, excess urination, and excess thirst). A person will excrete high volumes of dilute urine regardless of how much water they are drinking. High sodium (hypernatremia) concentrations in the blood may develop if the person does not have access to water to drink. Diabetes insipidus occurs with ADH deficiency (central or hypothalamic diabetes insipidus) or decreased renal response to the action of ADH (nephrogenic diabetes insipidus). Major causes of hypothalamic diabetes insipidus include damage to the hypothalamus such as with neoplastic, granulomatous or infiltrative diseases, neurosurgery, or severe head injury which prevents productions of ADH. Pituitary stalk damage can also cause DI due to interruption of the hypothalamic-pituitary nerve tracks. Pituitary adenomas do not typically cause DI, as tumor growth rarely affects the hypothalamus or the neurons involved in secreting ADH. Some patients have an idiopathic etiology (a diagnosis made only after exclusion of other possible causes of ADH deficiency). Idiopathic central diabetes insipidus is rare (< 0.005% of general population). Acquired nephrogenic diabetes insipidus may present in patients on long term lithium therapy, e.g. for bipolar disorder.
Diagnosis and management of DI
The diagnosis of diabetes insipidus should be suspected in patients with polyuria (frequent, high-volume urination). Patients may present with hypernatremia (high blood sodium levels) if they cannot drink enough water to keep up with water loss from the polyuria. Osmotic diuresis (e.g., glucosuria), hypokalemia and hypercalcemia are also causes of polyuria and should be excluded before making the diagnosis of diabetes insipidus. It is also important to exclude primary polydipsia, a condition characterized by increased water intake, most often seen in patients with psychiatric illness.
The diagnosis of diabetes insipidus requires the measurement of serum and urine osmoles when the patient is hypernatremic. With DI, the urine will be inappropriately dilute for the level of hyperosmolarity (concentrated blood, dilute urine). The diagnosis often requires a water restriction test. The normal response to fluid restriction (in the absence of DI) is an increase in plasma osmolality, which stimulates an increase in ADH release and subsequent increase in urine osmolality due to reabsorption of free water. This response is impaired in DI, and the patient is unable to concentrate their urine. In this test, the patient is advised to stop drinking water 2-3 hours before the start of the test. Serum and urine osmolality are measured every hour to monitor response. The test is continued until one of the following end points is reached:
- Urine osmolality normalizes. This indicates a normal ADH response to decreased fluid intake and suggests patient does not have DI.
- Urine osmolality remains stable over 2-3 hours, despite increase in serum osmolality suggesting impaired ADH release or action.
- Plasma osmolality increases to >300 mosmol/kg or plasma sodium >145 meq/L, also suggesting impaired ADH release or action.
If results of the water deprivation test are abnormal, the patient is given a dose of desmopressin (synthetic ADH) and urine osmolality is monitored. With central DI or ADH deficiency, administration of desmopressin should result in appropriate concentration of urine given the elevated serum osmolality. There would be no response with nephrogenic DI, as this represents ADH resistance. Results of this test for the different causes of polyuria are listed in Table 7.
|
|
Urine osmolality |
Serum osmolality |
Response to desmopressin |
|
Central DI |
No increase |
Increases |
Increase in urine osmolality |
|
Nephrogenic DI |
No increase |
Increases |
No effect |
|
Primary polydipsia |
Increases |
Increases |
N/A |
It is essential to provide adequate water intake to patients with diabetes insipidus, to replace the free water lost in the urine and avoid developing hypernatremia. Patients should be advised to drink based on their thirst. Patients with hypothalamic diabetes insipidus may also be treated with exogenous ADH therapy (desmopressin, Figure 12), which can help control their polyuria. Desmopressin is synthetic ADH and acts as a vasopressin V2R agonist. This leads to increased levels of apical aquaporin channels and increased water reabsorption.
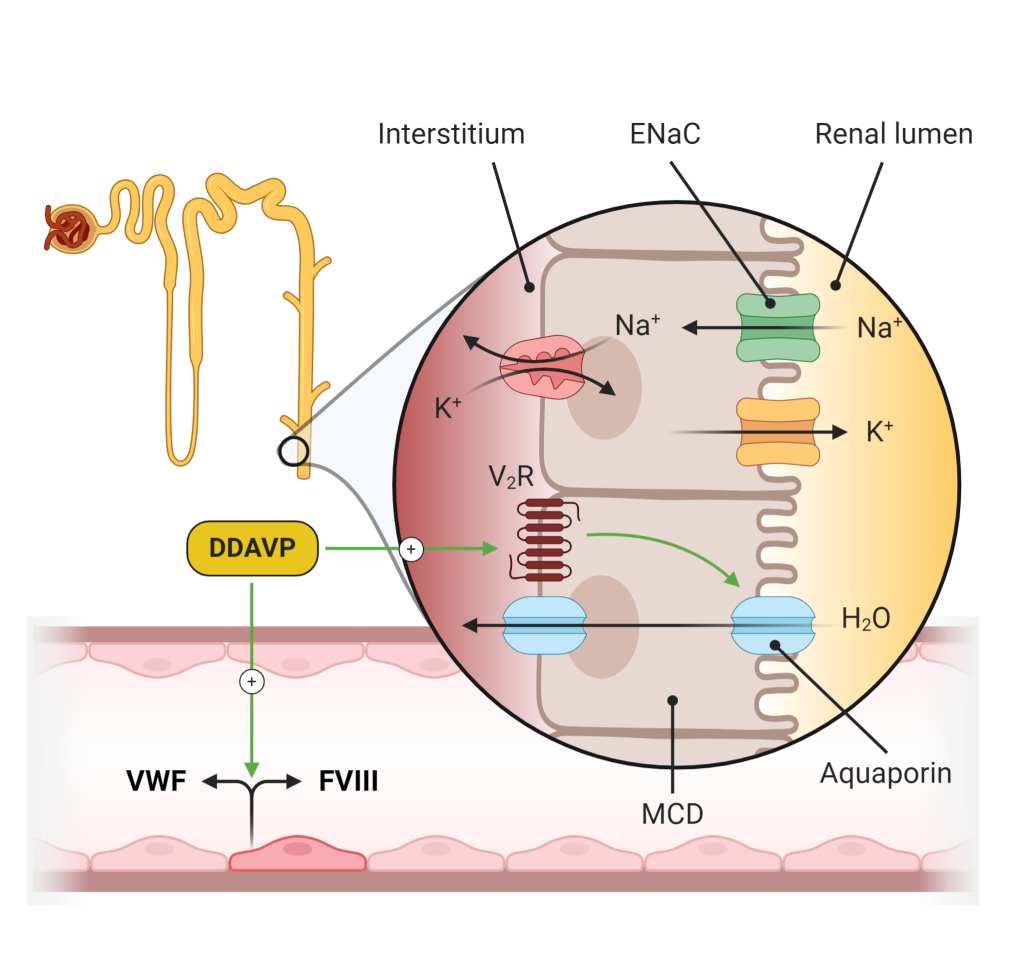
Syndrome of Inappropriate ADH (SIADH)
SIADH is a condition in which the hypothalamus synthesizes too much or the posterior pituitary releases too much ADH (vasopressin). This condition is the opposite of diabetes insipidus. When there is too much circulating ADH, the body is unable to excrete free water. Over time, the serum sodium levels decrease (hyponatremia) due to the dilutional effect of being unable to excrete free water. SIADH is a common condition that occurs after surgery on the pituitary gland. When it occurs, it is typically temporary. SIADH is one of many causes of hyponatremia. Pituitary surgery or trauma are only two of many causes of SIADH. Disorders of sodium and water will be discussed in more detail in your nephrology course.