
7 Diabetes Complications and Disorders of Lipid Metabolism
Introduction
In this chapter, we will focus on microvascular and macrovascular disorders that arise as a complication of type 1 or type 2 diabetes mellitus. This will include diagnosis and management of microvascular complications and macrovascular disorders such as hypertension and dyslipidemia. We will conclude by reviewing disorders of lipid metabolism.
Complications of Diabetes
Complications occur from both type 1 and type 2 diabetes mellitus. The complications reviewed here are seen in both conditions.
Treatment goals of diabetes are two-fold: 1) to prevent symptoms associated with hyperglycemia and 2) to prevent the long-term complications of chronic hyperglycemia. Diabetes mellitus is the leading cause of adult blindness, non-traumatic lower extremity amputation, and end stage renal disease. Patients with diabetes are 2 to 4 times more likely to develop coronary artery disease and heart disease is present in up to 20% of people with both type 1 and type 2 diabetes older than 45 years. Pregnancies in people with diabetes have an increased risk of complications.
Complications of diabetes are usually classified as: 1) macrovascular, involving the large blood vessels, including heart disease and cerebral vascular disease; 2) microvascular, those involving the small blood vessels; this includes diabetic retinopathy and nephropathy; and 3) neuropathic, which can involve the peripheral nervous system and/or autonomic nervous system.
Hyperglycemia, high blood pressure, and high cholesterol levels are known to damage the vascular endothelium. The damage to the vascular endothelium is a major contributor to the development of microvascular and macrovascular complications.
Macrovascular Complications
Macrovascular complications include diabetic cerebrovascular disease, diabetic cardiovascular disease, and diabetes peripheral arterial disease.
It is estimated that for patients with all types of diabetes, 75% of deaths are due to atherosclerosis (the process of cholesterol plaque buildup in the arteries which leads to narrowing). To prevent development (primary prevention) of atherosclerotic cardiovascular disease (ASCVD), it is crucial to aggressively manage cardiovascular risk factors such as lipid levels and blood pressure. Annual assessment of cardiovascular risk by using the ASCVD risk calculator from the American Heart Association will help guide treatment.
The central pathological mechanism in macrovascular disease is the process of atherosclerosis which leads to narrowing of arterial walls through the body. Atherosclerosis is the result of chronic inflammation and injury to the arterial wall in the peripheral or coronary vascular system. In response to this endothelial injury and inflammation, oxidized lipids from LDL particles accumulate in the endothelial wall of arteries. Monocytes then infiltrate the arterial wall and differentiate into macrophages, which accumulate oxidized lipids to form foam cells. Once formed, foam cells stimulate macrophage proliferation and attraction of T-lymphocytes. T-lymphocytes, in turn, induce smooth muscle proliferation in the arterial walls and collagen accumulation. The net result of the process is the formation of a lipid-rich atherosclerotic lesion with a fibrous cap. Rupture of this lesion leads to acute vascular infarction. Hypertension (high blood pressure), diabetes, and high cholesterol all increase the risk for development of atherosclerosis.
Part of managing blood pressure and cholesterol in a patient with diabetes is understanding that person’s risk for cardiovascular events in the future. Blood pressure should be checked at every visit. For individuals with a blood pressure >120/80 mmHg, lifestyle intervention is recommended. Lifestyle intervention consists of weight loss when indicated and a Dietary Approaches to Stop Hypertension (DASH)-style eating pattern, including reducing sodium and increasing potassium intake, moderation of alcohol intake, and increased physical activity. People with diabetes and blood pressure >130/80 should have a pharmacologic agent initiated to treat blood pressure with a treatment goal of <130/80. If albuminuria (urinary protein elevation) or coronary artery disease is present, an ACE inhibitor (eg. Lisinopril) or angiotensin receptor blocker (eg. Losartan) should be started. If no albuminuria or CAD is present, then any agent to treat high blood pressure can be initiated. Treatment goal is a blood pressure < 130/80.
Early and aggressive treatment of hyperlipidemia is an important part of minimizing cardiovascular risk in patients with diabetes. In fact, diabetes is considered a “cardiovascular disease equivalent” and essentially increases a patient’s risk of having a cardiac event to the same as if they had had a prior cardiac event. Elevated plasma LDL levels are associated with an increased risk of atherosclerosis and must be aggressively controlled for patients with diabetes to minimize their risk of cardiovascular disease. Lifestyle modification should focus on weight loss (if indicated), consumption of a Mediterranean and/or DASH eating pattern, reduction of saturated fat and trans fat, increase in omega-3 fatty acids and fiber, and increased physical activity. All of these lifestyle interventions can improve the lipid profile and reduce the risk of developing ASCVD in people with diabetes. Lipid panel and ASCVD 10-year risk should be assessed annually. For people with diabetes who are 40 – 75 years old, a moderate intensity statin (table 1) should be initiated with an LDL target of < 70mg/dL. For people with diabetes who are aged 40 – 75 years at higher cardiovascular risk (for example, diabetes and hypertension) it is recommended to use a high-intensity statin to target an LDL cholesterol goal of < 70 mg/dL (or <50mg/dL in some cases). For those with known ASCVD (regardless of age), a high intensity statin should be initiated. If LDL is >70 on high intensity statin, ezetimibe or PCSK9 inhibitor should be added. Statin medications are discussed in detail below.
Aspirin can also be considered for primary prevention for patients at intermediate or high risk of ASCVD (as determined by the risk calculator) with patient-shared decision-making but is generally not recommended in patients < 50 years of age.
For patients with established ASCVD or multiple risk factors for ASCVD, either an SGLT2-inhibitor (when eGFR > 20 mL/min/1.73m2) and/or a GLP-1 RA is recommended for reduction in major cardiovascular events or kidney events. In patients with heart failure, an SGLT2-inhibitor is recommended.
|
High intensity statin |
Moderate intensity statin |
|
Atorvastatin 40 – 80 mg |
Atorvastatin 10 – 20 mg |
|
Rosuvastatin 20 – 40 mg |
Rosuvastatin 5 – 10 mg |
|
|
Simvastatin 20 – 40mg |
|
|
Pravastatin 40 – 80mg |
|
|
Lovastatin 40mg |
|
|
Fluvastatin XL 80mg |
|
|
Pitavastatin 1 – 4mg |
Microvascular Complications
Dysfunction of vascular endothelium from hyperglycemia leads to the formation of microvascular complications. The pathophysiologic mechanisms leading to vascular endothelial dysfunction are described below. Microvascular complications include diabetic retinopathy, diabetic neuropathy, and diabetic kidney disease.
Potential pathophysiologic mechanisms of microvascular complications
The biochemical and molecular basis of the major chronic complications is not completely understood, but recent data have established the fundamental role of hyperglycemia. Multiple studies have shown that for individuals with both type 1 and type 2 diabetes, intensive diabetes management is beneficial for both preventing the development and delaying the progression of retinopathy, nephropathy, and neuropathy. Several biochemical pathways have been described, which may provide the mechanisms linking the end organ complications to the metabolic abnormalities of diabetes.
Nonenzymatic protein glycation
A considerable body of evidence has implicated the formation of advanced glycation end products (AGEs) via nonenzymatic protein glycation as an important mechanism for many of the complications present with diabetes. The best example of this is the nonenzymatic glycation of hemoglobin to form hemoglobin A1C (HbA1c). The formation of HbA1c provides clinicians and their patients the opportunity to assess overall diabetes control for the life of the hemoglobin molecule, usually about 90 days.
Nonenzymatic glycation occurs by a sequence of reactions in which glucose or other monosaccharides attach covalently to lysine and arginine residues within polypeptide chains to form glycation protein derivatives termed “Amadori products”. The ketoamines formed are effectively irreversible under physiologic conditions. AGE bind to receptors on inflammatory cells, endothelium, and vascular smooth muscle and cause cytokine and growth factor release, generation of reactive oxygen species, increase procoagulant activity of the endothelial cells and macrophages and increase proliferation of vascular smooth muscle cells and increased extracellular matrix (ECM) synthesis. In addition, AGEs crosslink with ECM proteins and further lead to endothelial injury from shear stress in all vascular endothelium and damage to the renal basement membrane. AGEs are involved in the development of macrovascular disease and and microvascular disease.
Polyol pathway
The sorbitol theory has been the most intensively studied. This postulate is based on the finding that glucose can be converted to sorbitol in most cells by the enzyme aldose reductase (Figure 1). With normal glycemia, intracellular sorbitol levels are low. During hyperglycemic states, excess glucose is shunted from the hexokinase/glycolysis pathway into other metabolic pathways. Cells containing aldose reductase (schwann cells, lense and retina, kidneys) convert glucose to sorbitol. Intracellular sorbitol increases with hyperglycemia. Once formed, sorbitol can be metabolized by sorbitol dehydrogenase into fructose. Some particularly sensitive organs lack sorbitol dehydrogenase. Fructose formation is slow, so sorbitol accumulates in the lens of the eye, peripheral nerves, retina, arterial and endothelial cells. Because sorbitol does not diffuse across cell membranes easily, it may accumulate and cause osmotic changes that damage the cell.

Diabetic Retinopathy
Diabetic retinopathy is the most common microvascular complication of diabetes. It is responsible for ~10,000 new cases of blindness every year in the United States. Risk for diabetic retinopathy increases with both duration of diabetes and severity of hyperglycemia. Hypertension also increases the risk for developing diabetic retinopathy.
Retinopathy may be the presenting sign in people with type 2 diabetes and can be present for many years prior to the diagnosis of type 2 diabetes. For that reason, screening for retinopathy is indicated when type 2 diabetes is diagnosed. For patients with diabetes without a known diagnosis of diabetic retinopathy, screening should occur annually with a dilated retinal exam. The first sign of diabetic retinopathy is nonperfusion of retinal capillaries, resulting in weakness of the capillary wall. The first signs of diabetic retinopathy may occur in childhood therefore screening should occur early. These changes are termed microaneurysms and are the classical finding in the early form of diabetic retinopathy, non-proliferative diabetic retinopathy (NPDR). More severe NPDR involves ruptured microaneurysms and leaking capillaries with intraretinal hemorrhages. Proliferative retinopathy (PDR), more advanced than the non-proliferative form, refers to new vessel formation. Blindness from diabetic retinopathy usually results from non-resolving vitreous hemorrhage, traction retinal detachment from the new vessel formation, or diabetic macular edema. (Figure 2).
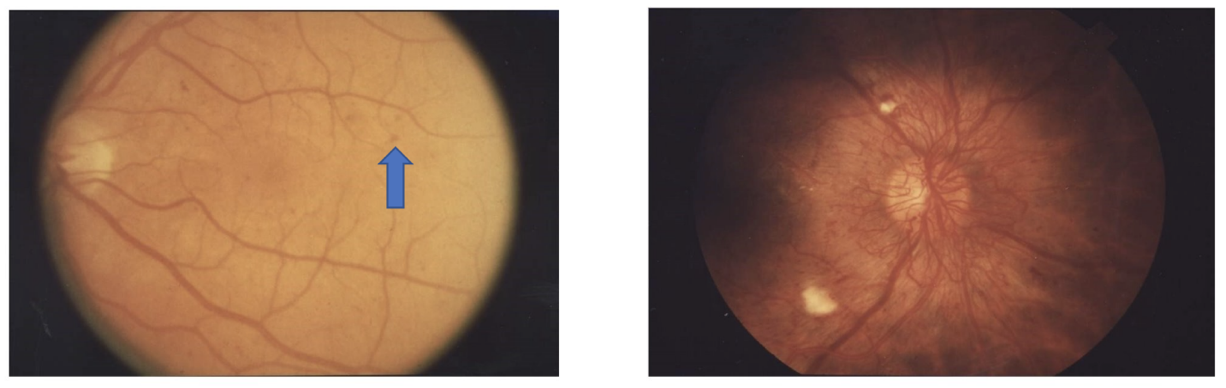
Management of blood sugars is the key to the prevention of diabetic retinopathy and slowing the progression once it occurs. Even an average blood sugar improvement of just 50 mg/dL can reduce the risk of developing retinopathy by 35% and slow the progression of existing retinopathy. For patients with established vision complications from their diabetes, there are several treatments available.
Treatment with pan-retinal laser can decrease the risk of severe visual loss from proliferative retinopathy to less than 5% and laser for clinically significant macular edema can decrease visual loss by greater than 50%, to about 12%. VEGF inhibitors have shown improvement in visual acuity in those with diabetic macular edema. It is due to these successful treatments that current recommendations are yearly dilated retinal exams for all adults with diabetes, to allow for early intervention and prevention of blindness.
Diabetic kidney disease
Diabetic kidney disease is defined as either reduced eGFR or albuminuria, or both without other apparent causes. End stage renal disease (ESRD) is a major public health problem for patients with diabetes, particularly those with type 2 diabetes, partly because etiology is often complicated by pre-existing hypertension. At least annually, urinary albumin (e.g., spot urinary albumin-to-creatinine ratio) and eGFR should be assessed in people with type 1 diabetes with duration of >5 years and in all people with type 2 diabetes regardless of treatment. In people with established diabetic kidney disease, the urinary albumin and eGFR should be monitored 1-4 times per year based on the severity.
Pathologically, in diabetic kidney disease (DKD) one sees glomerular basement membrane thickening and mesangial matrix expansion. A nodular lesion, pathognomonic for diabetic nephropathy, was first described by Kimmelsteil and Wilson in 1936. These glomerular changes are thought to be responsible for the glomerular hypertension seen in diabetic nephropathy. This leads to proteinuria, the hallmark of diabetic nephropathy. Eventually, systemic hypertension occurs. After the development of “dip-stick positive” proteinuria (when protein is detected on a laboratory dipstick which is about 500 mg/day), end-stage renal disease will occur in 3 to 5 years if left untreated.
Fortunately, aggressive treatment of the hypertension, especially with the use of angiotensin converting enzyme inhibitors (ACEI) and angiotensin receptor blockers (ARBs), can dramatically slow the progression of the renal disease. It is now appreciated that ACE inhibitors and ARBs are beneficial for patients with proteinuria, even without systemic hypertension. Either an ACE inhibitor or an angiotensin receptor blocker is recommended for those with elevated urinary albumin to creatinine ratio (>30 mg/g) in both type 1 and type 2 diabetes regardless of blood pressure. The renin-angiotensin-aldosterone system (RAAS) is known to be dysregulated in people with chronic kidney disease (CKD). CKD causes continuous overstimulation of the RAAS, leading to increased glomerular capillary pressure and activation of proinflammatory pathways that cause glomerular hypertrophy. Renin-angiotensin system blockade with ACEI and ARB decreases the intraglomerular pressure and slows the progression of kidney disease.
SGLT-2 inhibitors are also helpful in preventing the progression of diabetic kidney disease (DKD) and should be initiated in people with type 2 diabetes (even if the person has already met glycemic targets) who have diabetic kidney disease. SGLT2 inhibitors cannot be used in people with an eGFR < 20 mL/min/1.73m2. GLP-1 receptor agonists also have positive renal benefits and should be considered in people with DKD.
Diabetic Neuropathy
Many patients do not manifest a single type of diabetic neuropathy but rather a mixture of neuropathic features dominated by one or another subtype. The most common type of diabetic neuropathy is sensory or sensorimotor distal polyneuropathy. Sensory neuropathy often leads to an insensate foot, vulnerable to trauma, skin breakdown, and eventually ulceration and infection. Limb amputation may be required. A greater motor component will lead to focal or generalized weakness. Autonomic neuropathy may lead to bladder dysfunction, orthostatic hypotension, gastroparesis, sexual dysfunction, and diarrhea or constipation due to bowel dysmotility. Foot exams at every clinic visit are important to identify early signs of skin breakdown for patients with neuropathy, to avoid severe diabetic foot ulcers. For those with known peripheral neuropathy, foot exams should be performed every visit. For those without known diabetic neuropathy, foot exams should be performed annually.
Lipid Metabolism Introduction
Disorders of lipoprotein metabolism are clinically important due to their contribution to atherosclerotic cardiovascular disease. Diabetes mellitus and dyslipidemia, along with other factors, are major contributors which have led to the current epidemic of cardiovascular disease in the United States. Most of the content in this section is a review of what has been covered in previous blocks.
The term “lipids” refers to cholesterol (both free and esterified) and triglycerides (three fatty acids attached to a glycerol backbone) in the blood. Cholesterol has several important functions in the body: 1) it is a structural component of cell membranes; 2) it is a precursor for steroid hormones and vitamin D; and 3) it is also a precursor in bile acid synthesis. The major functions of triglyceride (TG) are: 1) as a long-term form of energy storage in fat; and 2) as an energy source for muscle in fasting and during exercise and/or for ketone formation after liberation of fatty acids.
Lipoproteins are complexes of proteins and lipids that are essential for the transport of cholesterol, triglycerides and fat-soluble vitamins.
Lipoprotein Structure and Function
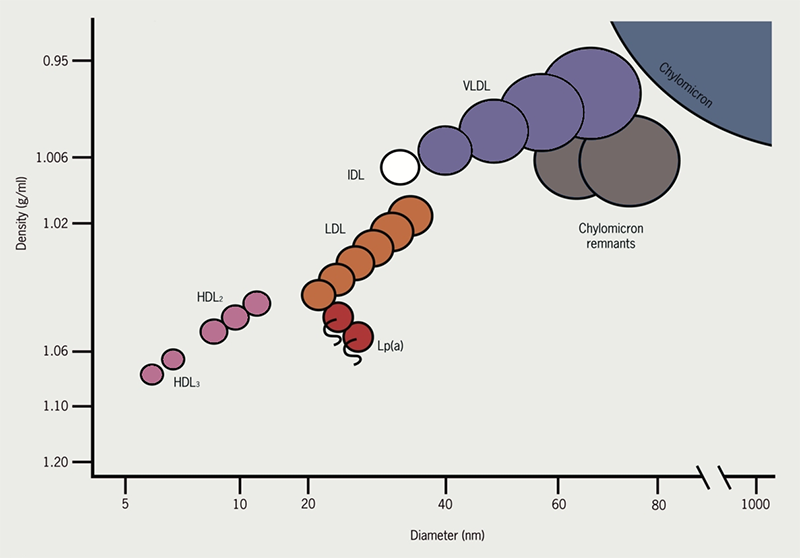
Neutral lipids such as cholesterol and triglycerides (TGs) cannot dissolve in body fluids such as plasma and lymph (since they are predominantly water); therefore, they are made miscible by incorporation into lipoproteins. Lipoproteins are large, mostly spherical complexes which carry lipids to and from various tissues. In other words, they transport cholesterol and triglycerides to peripheral tissues like liver, muscle and fat from the site of origin. Based on their buoyancy characteristics, lipoproteins are described by convention as chylomicrons (CM), very low-density lipoproteins (VLDL), intermediate density lipoproteins (IDL), low-density lipoproteins (LDL) and high-density lipoproteins (HDL) (Figure 3).
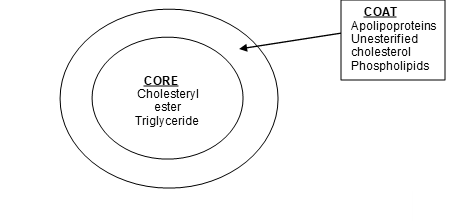
A mature lipoprotein particle is a sphere consisting of a central core of lipids (triglycerides and cholesteryl esters) surrounded by a surface layer of phospholipids, unesterified cholesterol, and proteins (apolipoproteins) (Fig 4).
Major Lipoprotein Classes
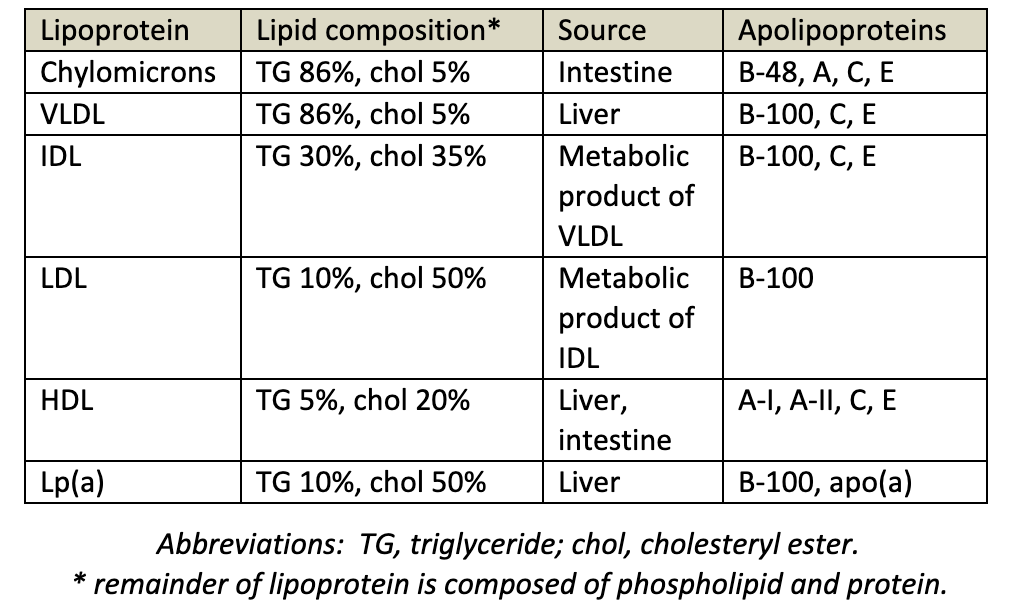
The major lipoprotein classes are chylomicrons, VLDL, IDL, LDL, HDL. Cholesterol is at the core of lipoproteins and is needed to maintain cell membrane integrity and synthesize bile acid, steroids, and vitamin D. Lipoproteins are summarized in table 2.
Chylomicrons deliver dietary TGs to peripheral tissues. They deliver cholesterol to liver in the form of chylomicron remnants, which are mostly depleted of their TGs. Chylomicrons are secreted by intestinal epithelial cells.
VLDL deliver hepatic TGs to peripheral tissue. They are the precursor for IDL and LDL. VLDL is secreted and synthesized by liver.
IDL are formed in the degradation of VLDL (when triglycerides in VLDL is hydrolyzed by LPL). IDL deliver TGs and cholesterol to liver.
LDL results from hepatic processing of IDL. LDL deliver hepatic cholesterol to peripheral tissues. The LDL core is rich in cholesterol ester and accounts for most of the cholesterol circulating in the blood during fasting. This lipoprotein plays a major role in the promotion of atherosclerosis.
HDL mediates reverse cholesterol transport from periphery to liver. Acts as a repository for apolipoproteins C and E (which are needed for chylomicron and VLDL metabolism). HDL is secreted from both liver and intestine. It also plays an opposite role to LDL in atherosclerosis by aiding in removal of tissue cholesterol by “reverse cholesterol transport.”
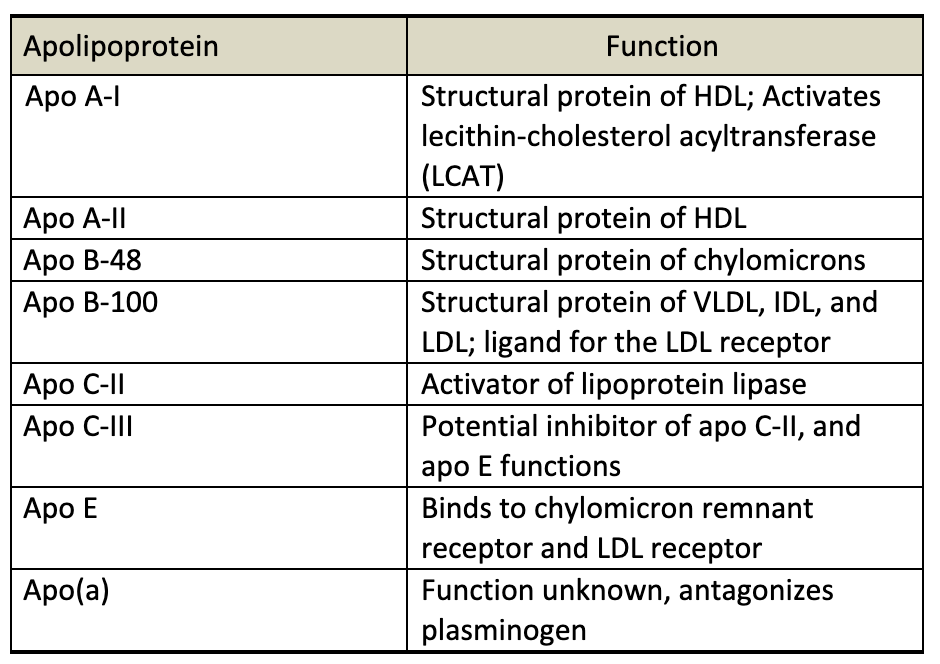
Apolipoproteins
Apolipoproteins are the protein component of lipids. They play several important roles in lipid metabolism. Apolipoproteins are involved in structure and assembly of the lipoprotein particle, they act as ligands mediating binding of lipoproteins to cell surface receptors. Apolipoproteins can activate enzymes important in lipoprotein metabolism. The apolipoproteins are listed below in table 3.
Key Plasma Lipid Enzymes
There are several enzymes critical to the functioning and processing of lipoproteins. These enzymes are listed below.
Lipoprotein lipase (LPL) causes the degradation of TGs circulating in chylomicrons and VLDS. Found on vascular endothelial surface. People with impaired lipoprotein lipase activity have higher levels of triglycerides (due to decreased clearance) and lower levels of HDL (due to reduced production from triglyceride-depleted VLDL remnants).
Hepatic lipase (HL) remove triglycerides and phospholipids from chylomicrons and VLDL remnants. HL also removes phospholipids from LDL and HDL. High hepatic lipase activity can result in small, dense LDL particles and smaller HDL particles. Small, dense LDL particles increase the risk of coronary artery disease.
Hormone-sensitive lipase (HSL) leads to the degradation of TGs stored in adipocytes. HSL is plays a pivotal role in providing the major source of energy for most tissues.
Lecithin-cholesterol acyltransferase (LCAT) converts cholesterol in the HDL particle to cholesteryl ester. This allows HDL to absorb more free cholesterol onto their surface from cells allowing the nascent HDL to become a mature HDL. LCAT deficiency leads to very low levels of HDL.
Cholesteryl ester transfer protein (CETP) mediates the exchange of triglycerides in VLDL, chylomicrons, and remnants for cholesteryl esters in HDL and LDL. Lower CETP levels promote HDL formation. Since higher HDL levels are associated with decreased risk of atherosclerosis, higher CETP activity is thought to lead to atherosclerosis.
Pathways of Lipoprotein Metabolism
Plasma cholesterol concentrations are maintained by biosynthesis through the endogenous pathway and absorption of dietary and biliary cholesterol through the exogenous pathway. In the endogenous pathway, cholesterol is synthesized by the liver and extrahepatic tissues and secreted into plasma, whereas the intestine is the primary site of the exogenous pathway of dietary cholesterol uptake. Alteration in either pathway will affect the concentration of plasma cholesterol.
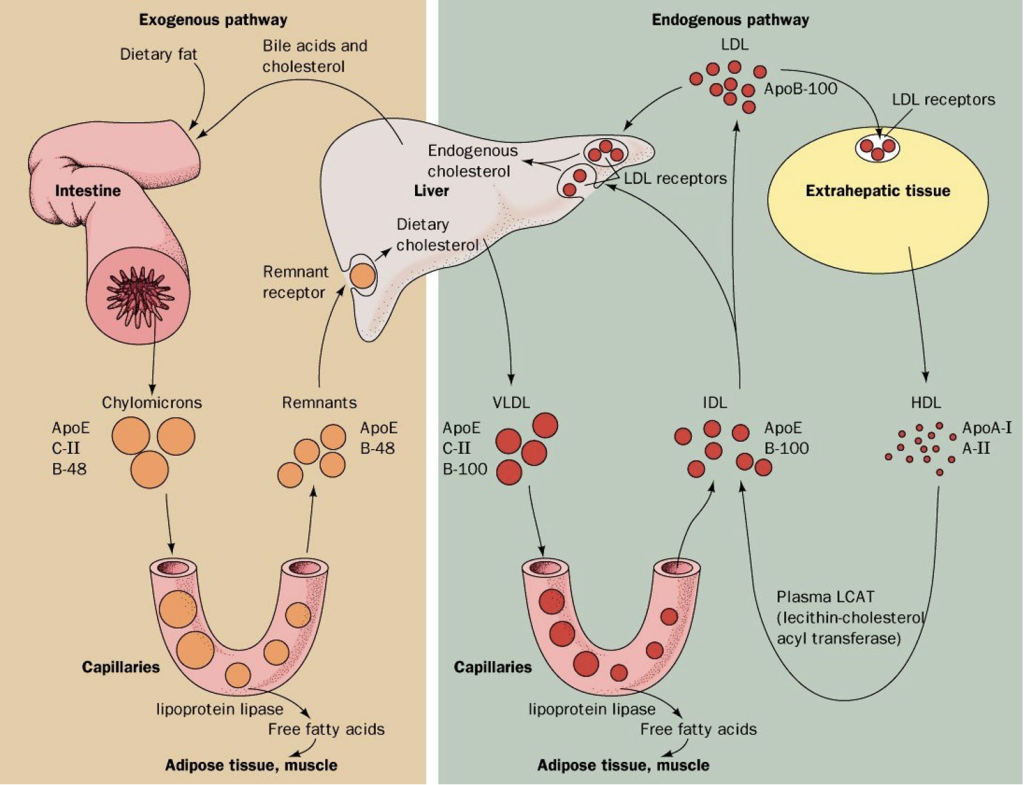
Exogenous Pathway
The process of fat from the diet is called the exogenous pathway of lipid metabolism. After a meal, dietary cholesterol and free fatty acids are absorbed into the epithelial cell, triglycerides are formed (from glycerol and free fatty acid) and triglycerides and cholesterol are assembled with Apo B-48 into chylomicrons (Fig 5). The chylomicrons travel in the lymphatics (thereby avoiding portal circulation) and the triglycerides are removed in the peripheral tissues. The remnant chylomicrons encounter lipoprotein lipase (LPL) on the capillary wall which hydrolyzes the triglycerides in the core of the chylomicron particle releasing free fatty acids (FFA). The free fatty acids are taken up by adjacent muscle cells or adipocytes. With the help of HDL, the remnant chylomicron is degraded by the liver which results in delivery of dietary cholesterol to the liver.
Endogenous Pathway
The hepatic secretion and metabolism of VLDL to IDL and LDL is referred to as the endogenous pathway of lipoprotein metabolism (Fig 5). VLDL are synthesized and secreted by the liver. These resemble the chylomicrons seen in the exogenous pathway, but VLDL have a different apolipoprotein (apo B-100 instead of apo B-48). Like chylomicrons, VLDL interacts with lipoprotein lipase (LPL) in capillary endothelium, and the core triglycerides are hydrolyzed to provide fatty acids to adipose and muscle tissues. The VLDL remnants are degraded to both IDL and LDL. LDL particles are cleared by the liver by the LDL receptor which binds to the apo B-100 on the LDL particles. The cholesterol from the LDL is used in synthetic actions (steroids, membranes, etc).
Causes of Dyslipidemia
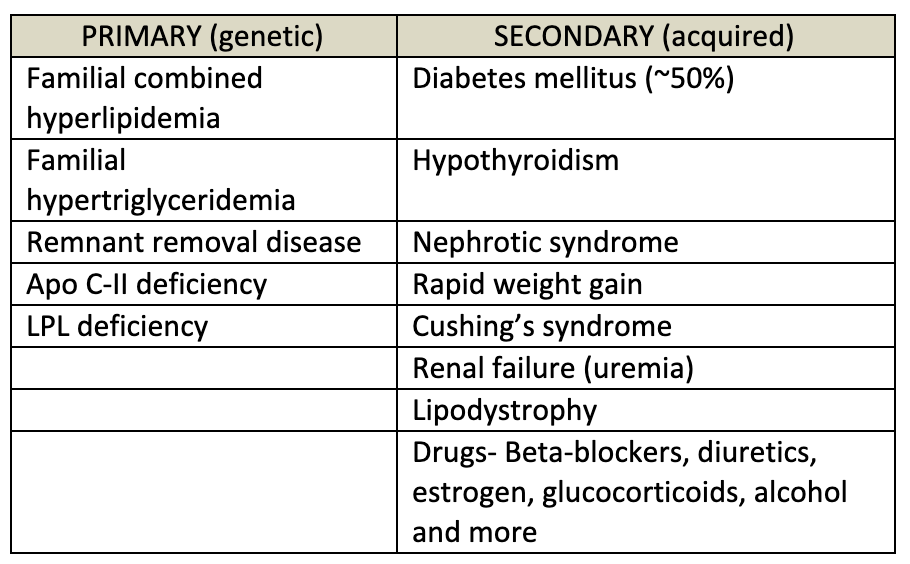
There are multiple causes (both environmental and genetic) that can lead to hyperlipidemia or hypolipidemia. Lipoprotein assembly, binding or breakdown can be affected; the enzymes that process lipids can be dysfunctional; several drugs and disease can cause secondary hypertriglyceridemia or hyperlipidemia; and environmental factors such as nutrient composition can also affect lipid levels.
When considering the cause of abnormal lipid levels, one should first consider which lipid level is abnormal (LDL, HDL, triglycerides, or several). Next, take care to evaluate for secondary causes of dyslipidemia such as diet, disease state, or certain medications. Secondary causes of dyslipidemia should be considered if the abnormality was recently diagnosed or if levels are significantly different from previously. Genetic causes should be considered, especially if there is a strong family history of dyslipidemia or early onset coronary artery disease. Genetic conditions are described below.
Most commonly, the cause of dyslipidemia is multifactorial, meaning it is due to a combination of polygenic inheritance, lifestyle, medications, and other disease states. The causes of acquired (secondary) hypertriglyceridemia are detailed in table 6 below. Monogenic causes of dyslipidemia (detailed below) can go undiagnosed for many years and may present when other factors (lifestyle, medications, other comorbidities) reveal the underlying abnormality.
Abetalipoproteinemia and hypobetalipoproteinemia are a genetic, autosomal recessive cause of low or absent LDL. The abnormality is an absence of apo B (abetalipoproteinemia) or deficiency of apo B (hypobetalipoproteinemia). Depending on the mutation, chylomicrons, VLDL, LDL are reduced or absent. People with abetalipoproteinemia do not have lipoproteins that contain Apo B-100 or Apo B-48 therefore numbers chylomicrons, VLDL, IDL, and LDL are absent. Clinical manifestations result from defects in the absorption and transport of fat-soluble vitamins. Affected individuals are deficient in vitamin A, D, E, K and develop neurologic defects. Children with abetalipoproteinemia have failure to thrive.
Familial hypercholesterolemia is a genetic, autosomal dominant cause of very high LDL. The abnormality is absent or defective LDL receptors which leads to very high LDL levels. The severity depends on heterozygote (more common/less severe) vs. Homozygote (extremely rare/very severe). The clinical manifestations are accelerated atherosclerosis, tendon xanthomas (cholesterol deposits on tendons), and corneal arcus (deposition of lipid in the peripheral cornea).
Dysbetalipoprotenemia (aka Remnant Removal Disease) is genetic, autosomal recessive cause of elevated triglycerides and cholesterol. The abnormality is defective ApoE which leads to marked impairment of hepatic remnant lipoprotein uptake. The clinical syndrome is variable based on phenotype. This mutation typically leads to premature atherosclerosis, eruptive xanthomas, and xanthoma striatum palmare.
Familial hypertriglyceridemia is due to overproduction of triglycerides contained within a normal VLDL particle and is characterized by excessive amount of triglycerides. The molecular basis is not known. When this condition exists with other causes that are known to increase triglycerides (see table 6) severe hypertriglyceridemia can result.
Familial combined hyperlipidemia (FCH) is due to excessive amount of apo B which is secreted as VLDL and/or LDL particles. The VLDL are smaller than normal. The molecular basis not known. The difference from familial hypertriglyceridemia is that FCH leads to both high TG and high LDL. Due to the high LDL, there is a higher risk for cardiovascular disease. When this condition exists with other causes that are known to increase triglycerides (see table 6) severe hypertriglyceridemia can result.
Chylomicronemia is an abnormal persistence of chylomicrons in the plasma for 12-24 hours after a meal leading to severe hypertriglyceridemia (TG >1000mg/dL). It can occur when LPL is deficient or due to a genetic defect in apo C-II. Much more commonly, markedly high triglyceride levels are caused by the coexistence of a primary genetic form of hypertriglyceridemia (either familial hypertriglyceridemia or familial combined hyperlipidemia) combined with a secondary or acquired disorder of plasma triglyceride metabolism. Symptoms of severe hypertriglyceridemia include pancreatitis, eruptive xanthomas, reversible memory loss, and peripheral neuropathy. Plasma triglycerides are typically > 1000 mg/dL when this syndrome occurs.
Mutations in the enzymes and lipoproteins associated with HDL, causing alterations in HDL levels, are rare. Unlike the genetic hypercholesterolemia conditions (with high VLDL and LDL levels) which always result in premature atherosclerosis, low HDL-C is not always associated with accelerated atherosclerosis. The risk associated with low HDL levels depends upon the underlying cause. Mutations that affect the following proteins and enzymes will disrupt HDL metabolism: Apo A1, ABCA-1 (needed for HDL stability), LCAT deficiency (needed for HDL formation), CETP deficiency (very high HDL levels).
Management of Dyslipidemia
Treatment of abnormal lipid levels depends on the disorder that exists. Most frequently, you will be encountering elevated LDL and TG in your clinical practice. The goal of treatment of dyslipidemia (especially elevated LDL and TG) is to prevent the development of other diseases (particularly cardiovascular disease) that can occur from dyslipidemia. There is significant focus on medical management of dyslipidemia, but lifestyle modification is equally important and should be part of all treatment plans for dyslipidemia.
Lifestyle Modification
There are many dietary modifications that can be made with the goal of lowering LDL levels. Dietary saturated fatty acids decrease hepatic LDL receptor activity and lead to more circulating LDL. Conversely, monounsaturated and polyunsaturated fatty acids lower LDL levels by increasing the hepatic LDL receptor activity. The consumption of dietary cholesterol (egg yolks, shrimp, beef, cheese, butter, etc) do lead to increase in LDL levels but the effect is modest and typically reversible with limiting cholesterol intake. Trans fatty acids, found in fried foods and commercial baked goods, lead to increased LDL levels and should be avoided. Fiber lowers LDL levels by decreasing cholesterol absorption in the small intestine. When carbohydrates are consumed in excess, they provide a substrate for free fatty acid synthesis which leads to excess VLDL secretion leading to increased triglyceride levels.
With the above in mind, patients with elevated LDL should be advised to eat a diet with an emphasis on vegetables, fruits, legumes, nuts, whole grains, fiber and fish. Trans fats and dietary cholesterol intake should be limited. People who have severe hypertriglyceridemia (TG >1000 mg/dL) can decrease circulating triglyceride levels over 24 hours by eliminating dietary fat.
Other than diet, weight loss (when appropriate) can lead to lower LDL levels. Limiting alcohol intake is critical, especially to manage high triglyceride levels. Exercise is not effective in lowering LDL levels but does lead to increased HDL levels.
Drugs used to lower LDL levels
Treatment principles – Current guidelines for management of high blood cholesterol levels recommend statin treatment (discuss in the following section) for four groups of patients. In patients without known history of ASCVD the goal is primary prevention of cardiovascular disease (CVD). A risk calculator (for people 40 – 75 years old) designed by the American Heart Association (AHA) can be found here (https://tools.acc.org/ascvd-risk-estimator-plus/#!/calculate/estimate/). Need to start a cholesterol lowering medication (typically a “statin”) can be determine based on the AHA Risk Calculator.
Patients in whom cholesterol lowering therapy should be initiated:
- Patients with clinically evident atherosclerotic cardiovascular disease (ASCVD)
- Primary LDL-C levels of > 190 mg/dL
- Type 1 or type 2 diabetes, age 40-75, and an LDL-C level of > 70 mg/dL
- a 10-year risk of ASCVD of 7.5%, and an LDL-C level of > 70 mg/dL
For those with an LDL of <70 mg/dL, multiple agents may be required. It is possible that an individual may require a statin, a PCKS9i, and/or ezetimibe to achieve LDL target. Statin medications are always first line therapy for LDL lowering unless there is a contraindication to statin use.
Statins or HMG-CoA Reductase Inhibitors (Fig 6) are effective agents for lowering LDL cholesterol levels (20-60%). They raise HDL levels moderately (5-15%), and this effect appears to be greatest in those with elevated baseline triglycerides. They also may lower triglycerides, although this effect is variable and related to the baseline level of triglyceride, as well as the dose of the statin.
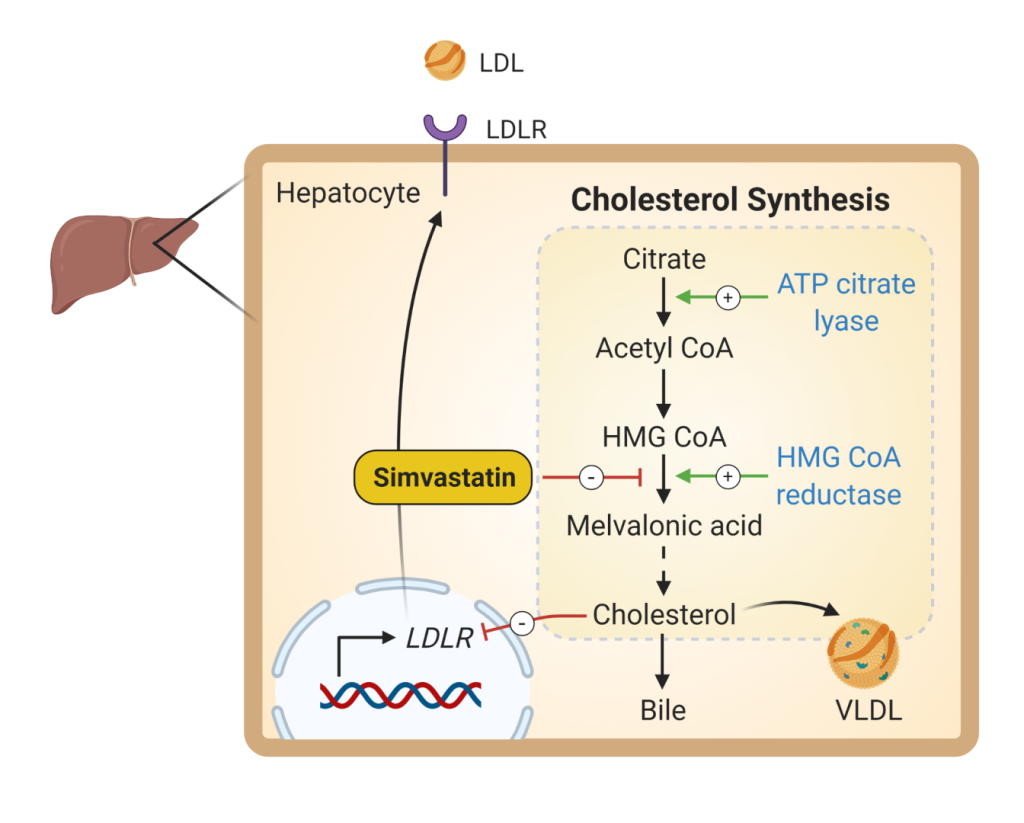
These agents lower cholesterol levels by a competitive inhibition of HMG-CoA reductase, the rate-limiting step in cholesterol biosynthesis. Statins may have a number of other effects (also called pleiotropic effects) independent of their cholesterol lowering abilities that mitigate the processes of atherosclerosis. There is evidence suggesting that statins reduce inflammation, improve endothelial dysfunction, and reduce thrombogenicity. However, the extent of LDL reduction appears to be the major determinant of cardiovascular risk reduction. Multiple primary and secondary intervention studies have demonstrated these agents to be highly effective in reducing cardiovascular events.
Overall, these agents are remarkably well tolerated. They produce reversible hepatic transaminase elevations in 0.1-2.5% of patients. Nonspecific myalgias are the most common side effect. Rarely, they may cause a severe myositis and muscle breakdown. Statins are useful for treatment of most major types of hyperlipidemia.
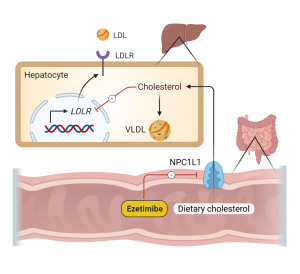
Ezetimibe is a potent intestinal cholesterol absorption inhibitor. It becomes localized in the intestinal brush borders where it inhibits Niemann Pick C1-Like 1 (membrane cholesterol transporter expressed in GI epithelial cells and hepatocytes). Inhibition of NPC1L1 decreases absorption of dietary and biliary cholesterol (Fig 7). It increases fecal cholesterol excretion. It does not affect absorption of triglycerides, fatty acids or fat-soluble vitamins across the brush border. Alone, it can reduce LDL levels 14-17%. When added to a statin it can provide a synergistic effect of an additional 25% reduction in LDL levels. This agent is generally very well tolerated but can sometimes cause GI disturbance and muscle pain.
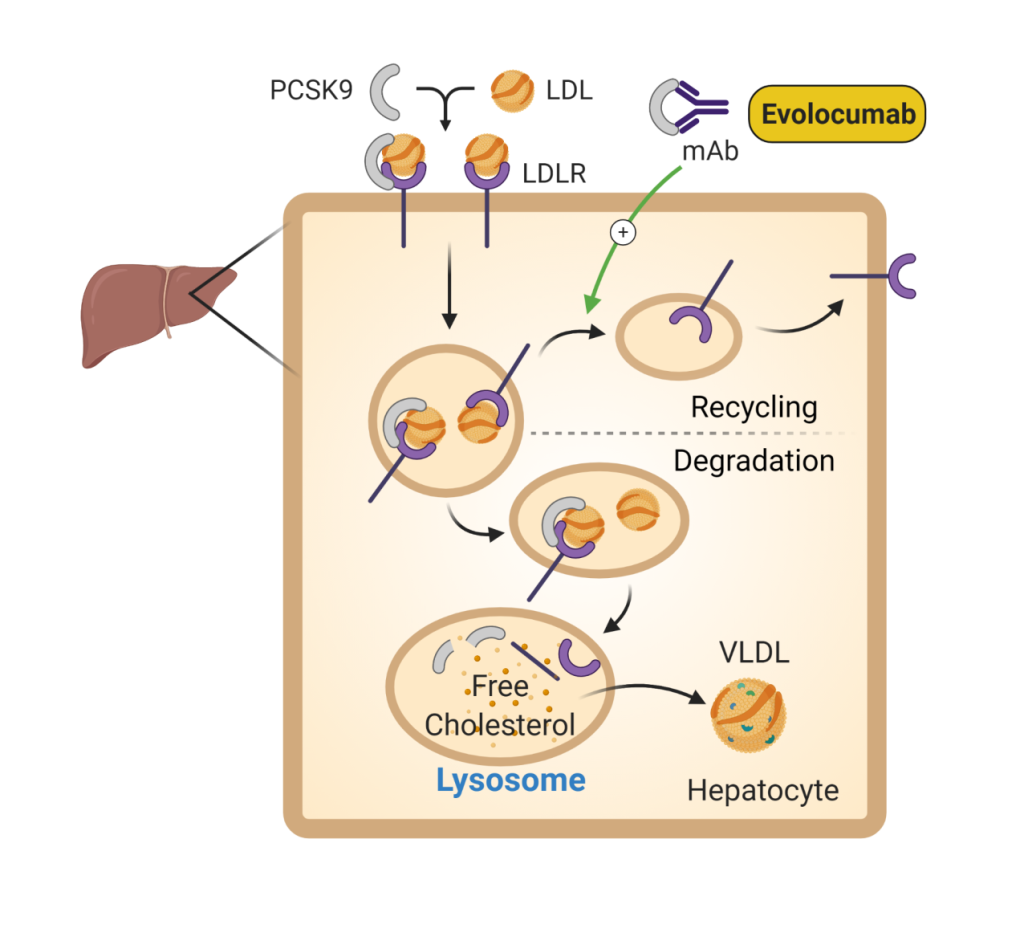
PCSK9 inhibitors (Fig 8) are one of the newest medications available for treatment of elevated LDL. PCSK9 is a protein that prevents recycling of the LDL receptor to the cell surface, decreasing the availability of LDL receptors to clear LDL from the circulation. PCSK9 inhibitors block the action of this protein, which results in more LDL receptors available to bind LDL and results in a dramatic lowering of LDL levels. Use of PCSK9 inhibitors has been shown to decrease cardiovascular events, but no significant effect on cardiovascular death in patients with established cardiovascular disease. Injection site reactions are the most common complication. At a monthly cost of over $14,000 this medication is cost-prohibitive for many patients.
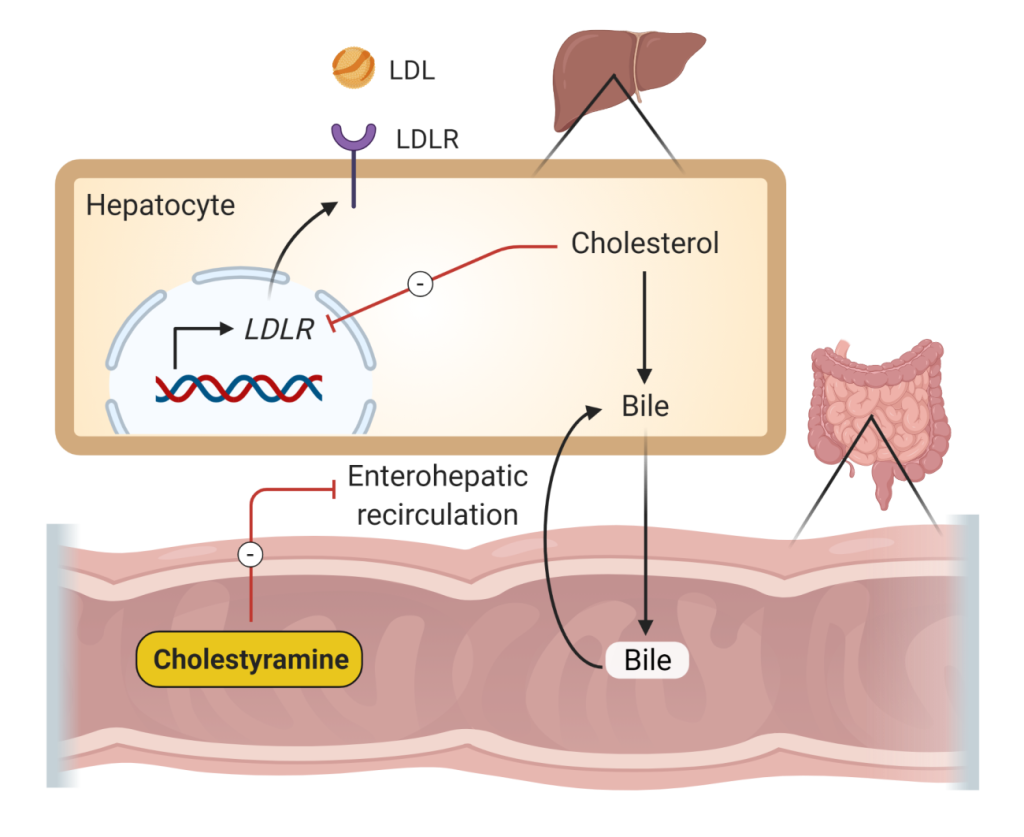
Bile acid sequestrants or resins lower LDL cholesterol levels (10-25%) and may raise HDL cholesterol levels modestly (3-5%). They may significantly raise triglyceride levels, particularly in those patients with baseline hypertriglyceridemia. Therefore, these agents should be used only in patients with pure hypercholesterolemia. These agents bind and sequester bile acids in the GI tract. The bile acids are not reabsorbed, which decreases enterohepatic reabsorption, increases hepatic cholesterol to bile conversion, decreases hepatic cholesterol levels, and increases LDL receptor expression, thus decreasing plasma LDL (Figure 9). Their use is limited by frequent gastrointestinal side effects (e.g. constipation, dyspepsia, reflux esophagitis).
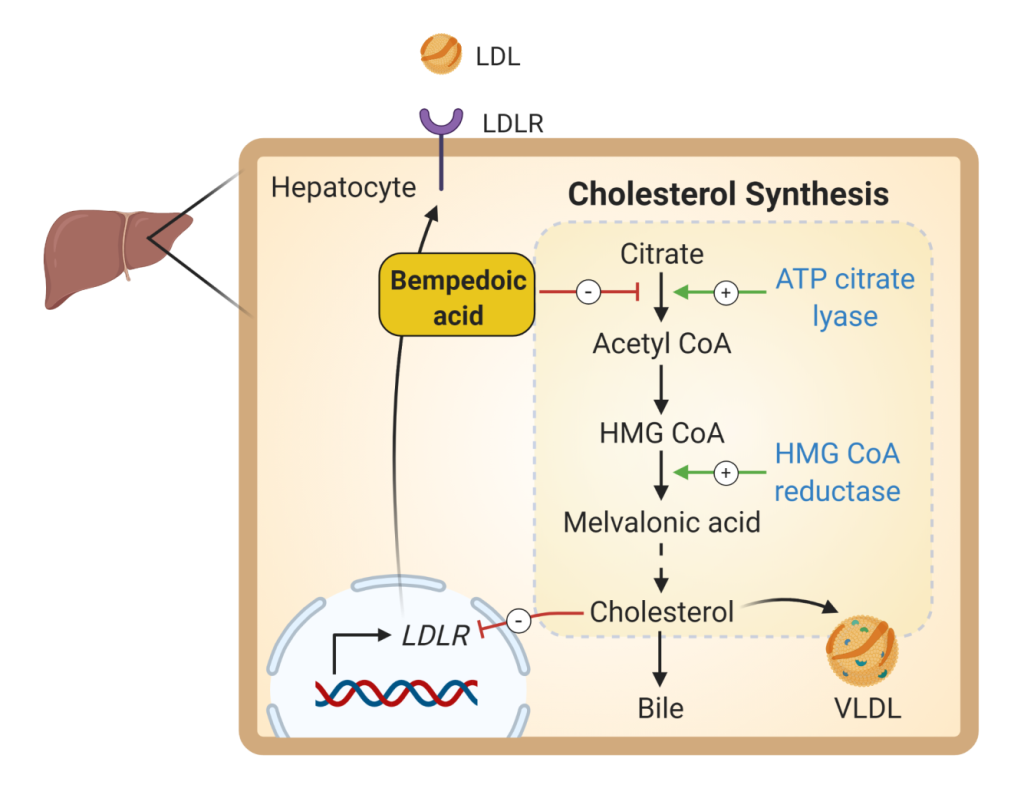
Bempedoic acid (Fig 10) is the newest cholesterol lower agent to come to market. It inhibits the enzyme ATP citrate lyase which is upstream of HMG CoA reductase. Similar to statin medications, bempedoic acid inhibits cholesterol biosynthesis. The main toxicity is LFT elevation. More mild side effects are similar to that of statins (eg. Myalgias). Bempedoic acid is currently FDA approved as an adjunct to diet and maximally tolerated statin therapy for the treatment of hyperlipidemia.
Drugs used to lower triglyceride levels
Medications to lower triglyceride levels should be initiated when triglyceride levels are greater than 500 – 1000 mg/dL (depending on other risk factors that may lead to elevated triglycerides, such as diabetes or alcohol use). The goal of treatment is to prevent the occurrence of pancreatitis which typically occurs when triglyceride levels are greater than 1000 mg/dL.
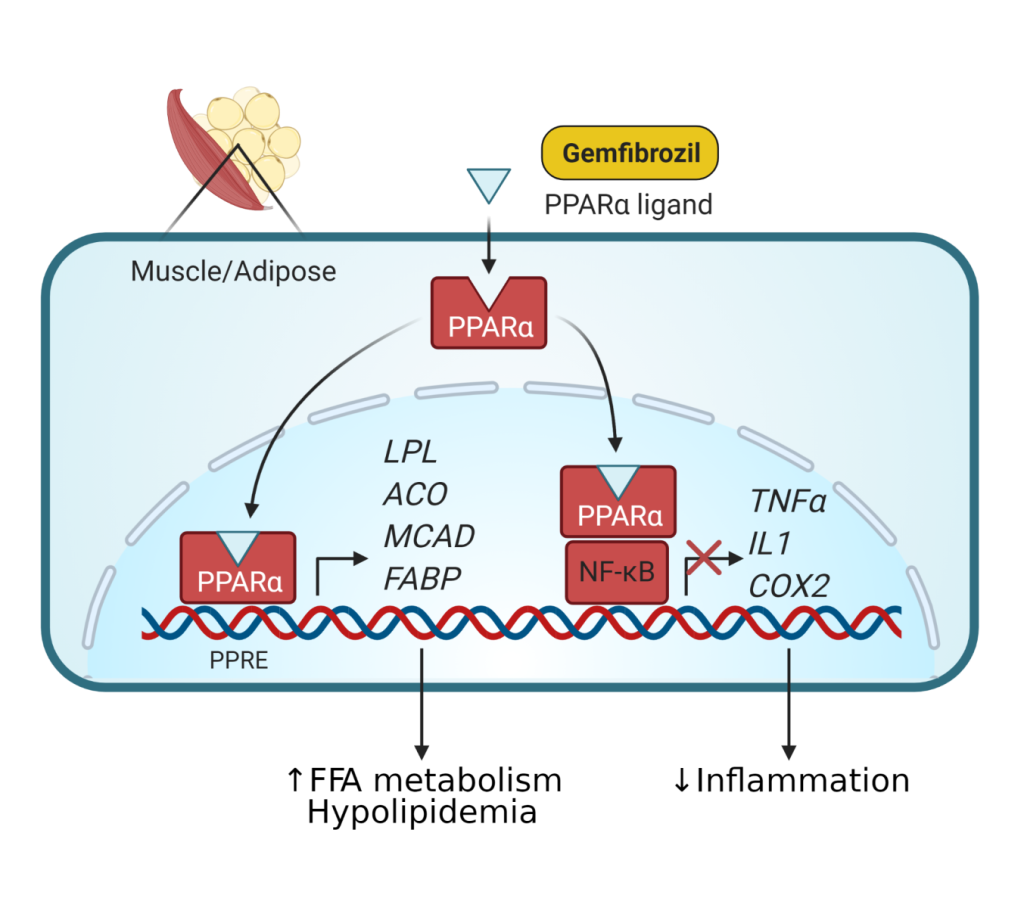
Fibrates are fibric acid derivatives (gemfibrozil and fenofibrate) and lower triglycerides (20-50%) and raise HDL cholesterol levels (10-30%). They are the most effective agents for lowering triglycerides. They have a variable effect on LDL cholesterol levels and can raise LDL levels in some hypertriglyceridemic patients. The newer fibrates (fenofibrate, bezafibrate) appear to have more beneficial LDL lowering effects.
The mechanism of action (Fig 11) is primarily by acting as a peroxisome proliferator activated receptor ⍺ (PPAR⍺) agonist which stimulates lipoprotein lipase activity and triglyceride hydrolysis. These agents are generally well tolerated but can produce a variety of gastrointestinal side effects. The main indication for using these drugs is in severe hypertriglyceridemia.
Niacin lowers LDL cholesterol (10-25%), and triglyceride levels (20-50%), while increasing HDL cholesterol levels (15-35%). It decreases small dense LDL particles. Because of these varied beneficial lipid effects, niacin is commonly a first or second-line agent in treating all dyslipidemias associated with cardiovascular disease.
The primary side effects are flushing and pruritus. It can cause hyperglycemia and worsen insulin resistance, and therefore its use can be problematic in some diabetic patients. At higher doses it can cause hepatotoxicity. It also, less commonly, causes gastrointestinal upset and raises uric acid levels and may therefore precipitate or aggravate gout. An extended-release formulation of niacin is reported to have lesser liver toxicity and flushing. Niacin is commonly used in mixed hyperlipidemia (with high cholesterol and triglycerides), usually in combination with a statin. Unfortunately, larger clinical trials studying the effect of niacin in combination with statin therapy have not shown significant cardiovascular benefit beyond statins alone.
Omega-3 fatty acids in high doses (>6 grams/day) can reduce triglycerides (25-30%) by inhibiting hepatic VLDL-TG synthesis without effects on LDL or HDL. Supplementation can modestly raise HDL. However, side effects such as nausea and fishy taste after burping can be distressing to many patients. There is no evidence for cardiovascular benefit with supplementation of fish oil in patients with diabetes. In general, omega-3 fish oil is not routinely prescribed for lipid lowering therapy.