
5 Calcium and Bone Mineral Metabolism
Introduction
Calcium is necessary for most cellular ‘actions’ – muscle contraction, neuronal firing, and hormone secretion, to name only a few. These processes all depend on intracellular calcium flux which, in turn, is partially determined by the serum concentration of ionized calcium. In neurons and myocytes, for example, a decrease in ionized serum calcium increases excitability; thus, low serum calcium levels can cause seizure, tetany, and cardiac arrhythmias. In contrast, an increase in ionized serum calcium decreases excitability. Clinical manifestations of hypercalcemia include coma and weakness, as well as cardiac arrhythmias distinct from those associated with hypocalcemia. In most cells the right amount of calcium is required for the cell’s metabolic machinery to function properly. Calcium is also important for bone health. As might then be expected, the concentration of calcium both inside and outside the cell is very tightly regulated through multiple mechanisms.
Calcium
Ionized calcium
Approximately 45% of calcium is present free in the circulation. The rest is bound to albumin and other anions. The regulation of calcium balance is primarily concerned with maintaining normal free, or ionized, calcium levels. Like hormones bound to carrier proteins, anything that affects albumin levels can lead to changes in total calcium levels, but ionized calcium levels remain stable. Changes in acid-base status can also shift the amount of calcium bound to albumin and alter ionized calcium levels. In conditions of alkalosis, with elevated pH, there is increased calcium binding to albumin. This leads to low ionized calcium. Acidosis, with low pH, decreases calcium binding and leads to increased ionized calcium.
Renal calcium handling
The main route of calcium excretion is through the kidney. The kidney reabsorbs almost all the calcium that is filtered daily. Renal excretion of calcium is essential to maintain calcium homeostasis, and therefore problems with renal function can have a large impact on serum calcium concentration.
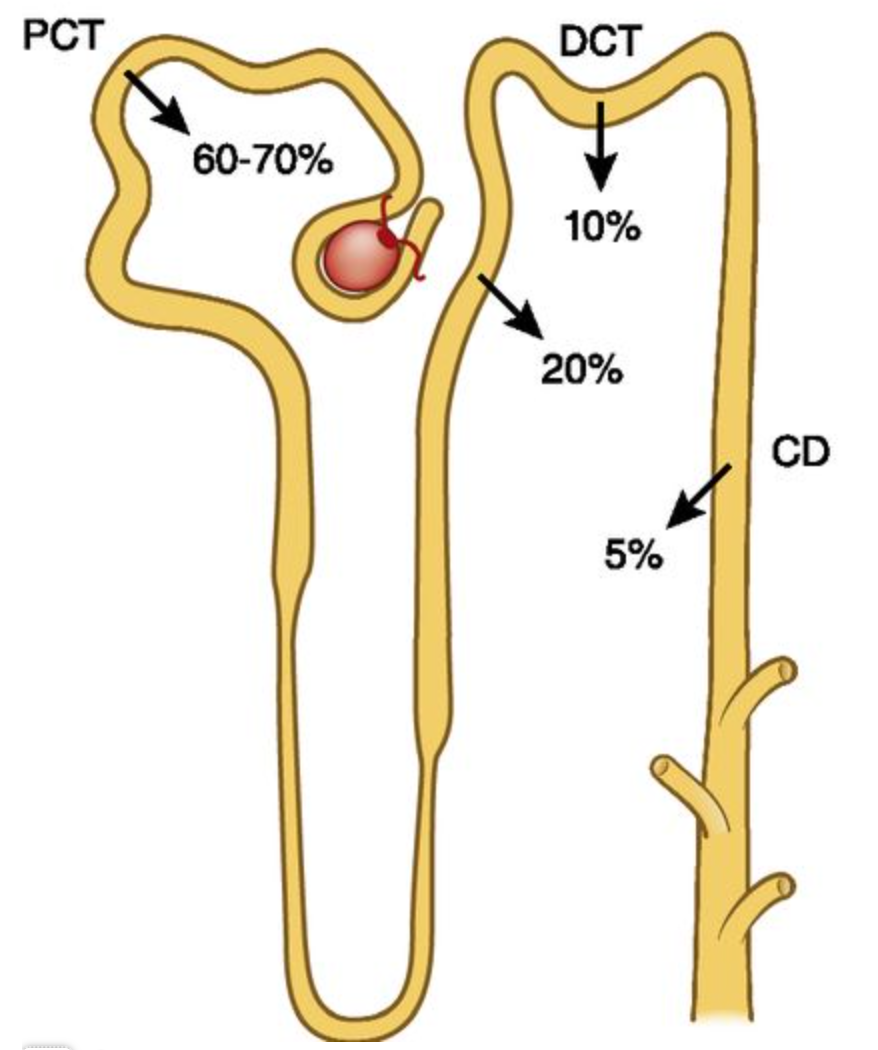
20% of calcium is resorbed at the thick ascending loop of Henle (TALH) (figure 1). The calcium sensing receptor (CaSR) is found in the TALH and senses serum calcium levels. If serum calcium is low, CaSR signaling diminishes and results in enhanced reabsorption of calcium from the urine. If serum calcium is high, CaSR signaling increases and more calcium is excreted in the urine.
Calcium reabsorption in the distal convoluted tubule (DCT) is regulated by parathyroid hormone. The details of this will be covered in a later section.
Bone and intestinal calcium absorption
An important element in the maintenance of normal calcium levels is the skeletal system. Bone is the main storage depot of calcium in the body, and mobilization of calcium from bone allows for the precise control of calcium levels that we normally see. The regulation of bone turnover will be discussed in a later section.
Dietary calcium is absorbed into the circulation from the gut. Generally, calcium is not well absorbed in the gut. There are two different mechanisms of absorption: transcellular (active) and paracellular (passive). The transcellular process is dependent on vitamin D. Vitamin D regulates synthesis of the proteins that mediate transport of calcium from the apical membrane of the enterocyte to the basolateral membrane. To a much lesser degree, vitamin D impacts the tight junctions that control paracellular transport of calcium. Because calcium absorption in the gut is dependent on vitamin D, low vitamin D levels can result in low calcium, due to decreased gut absorption. Conversely, high vitamin D levels may result in hypercalcemia due to excess absorption in the gut. Gut absorption of calcium is also inhibited by glucocorticoids, so patients on high dose steroids are at higher risk of complications related to low calcium.
Other factors that increase the secretion of PTH include low serum calcium, low serum 1,25-(OH)2-vitamin D (calcitriol) or a high serum phosphate (figure 2). High serum calcium, 1,25-(OH)2-vitamin D as well as low phosphate decrease PTH synthesis and release.
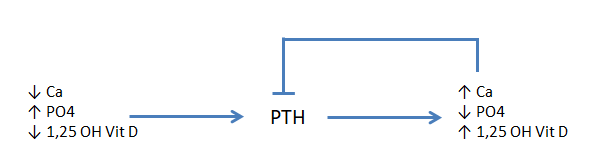
All the actions of PTH on mineral homeostasis are mediated through a G-protein coupled transmembrane receptor (PTH-1R). Binding to its receptor leads to activation of adenylate cyclase and generation of cAMP which leads to different effects depending on the target tissue. PTH increases the release of calcium and phosphate from bone. In the bone, PTH induces RANKL expression in osteoblasts which leads to activation of osteoclasts and increased bone turnover. PTH also induces IL-1 expression on osteoblasts which leads to activation of osteoclasts. In the kidney, PTH acts at the distal convoluted tubule to increase the number of specialized calcium channels at this site thereby increasing the amount of calcium reabsorbed. PTH also triggers decreased phosphate reabsorption, by down-regulation of a sodium/phosphate co-transporter. PTH stimulates 1,25-(OH)2-vitamin D production by the kidney through activation of the enzyme 1-α hydroxylase, which is necessary to convert inactive 25-OH-vitamin D (calcidiol or calcifediol) to the active form, 1,25-(OH)2-vitamin D (calcitriol) and therefore facilitate increased absorption of calcium at the gut.
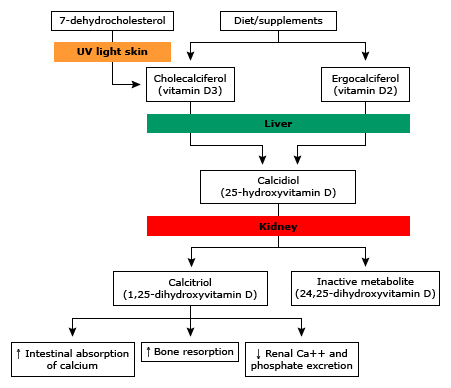
Vitamin D deficiency is common and generally results from a combination of low sunlight exposure, age, and inadequate dietary vitamin D. Symptoms are usually absent or insidious and non-specific, such as bony pain and neuromuscular weakness. Vitamin D deficiency can usually be corrected easily by oral replacement. When Vitamin D is low, this causes an appropriate rise in parathyroid hormone. This is a type of secondary hyperparathyroidism (more on this below).
Calcitonin
Calcitonin, a 32-amino-acid peptide hormone, remains a puzzle. Circulating calcitonin comes from the neural crest-derived parafollicular C-cells within the thyroid. Release of calcitonin from the C-cells is stimulated by high serum calcium levels through the CaSR. Low serum calcium suppresses calcitonin release. Acute changes in serum calcium have a much greater effect on calcitonin than chronic changes do. The effect of calcitonin on calcium homeostasis is to suppress osteoclast activity, inhibiting bone resorption. This effect is potent and rapid and is mediated by a specific calcitonin receptor, one of the few receptor types abundantly expressed on the surface of osteoclasts.
|
PTH-mediated |
Non-PTH mediated |
|
|
Hyperparathyroidism. Parathyroid hormone may be elevated appropriately (in response to abnormal vitamin D, calcium, or phosphorus levels, known as secondary hyperparathyroidism) or inappropriately. When the PTH is elevated inappropriately (without responding to correct signals) this often leads to hypercalcemia and is typically termed primary hyperparathyroidism (this pattern can be seen in tertiary hyperparathyroidism as well, see below). A single parathyroid adenoma is the most common cause of inappropriately elevated PTH secretion (primary hyperparathyroidism), and this is the most common diagnosis for patients in whom hypercalcemia is incidentally discovered. Four-gland hyperplasia can also cause primary hyperparathyroidism, either sporadically or in genetic conditions such as multiple endocrine neoplasia (MEN) type 1 or 2A. Parathyroid carcinomas are very rare but can cause hyperparathyroidism and often severe hypercalcemia.
Multiple endocrine neoplasia (MEN) causes primary hyperparathyroidism in type 1 or 2A. Familiarity with these conditions is essential for success on USMLE Step 1 and 2. MEN is a term used to describe three syndromes associated with certain hormone-producing neoplasms. MEN 1 is caused by a mutation in the menin gene and is associated with endocrine pancreatic tumors, primary hyperparathyroidism, and pituitary adenomas (3 “P”s). MEN2A and MEN2B are caused by a mutated RET proto-oncogene and both typically present with medullary thyroid cancer (MTC) and pheochromocytoma. MEN2A also is associated with primary hyperparathyroidism (1 “M”, 2 “P”s). MEN2B is associated with mucosal neuromas and marfanoid habitus in addition to pheochromocytoma and MTC (2 “M”, 1 “P”).
Patients with primary hyperparathyroidism are often asymptomatic. On labs, they have elevated calcium with elevated PTH and hypophosphatemia. Alkaline phosphatase (an enzyme that is a marker of bone turnover) is often elevated due to increased bone turnover. Classically, they have bone pain associated with “brown tumors”, which are cystic bone spaces filled with brown fibrous tissue. Despite high PTH levels, they often have elevated urinary calcium levels. The CaSR detects the high calcium levels in the blood and blocks calcium reabsorption from the urine. The high urinary calcium reaching the DCT overwhelms the kidneys’ ability to reabsorb calcium, leading to elevated urine calcium. This contributes to the development of renal stones seen with hyperparathyroidism.
Kidney disease, hypocalcemia, hyperphosphatemia and vitamin D deficiency can lead to elevated parathyroid hormone levels (secondary hyperparathyroidism). This PTH elevation is in response to abnormal phosphorus, calcium, or vitamin D and is increased with the goal of maintaining normal calcium (in vitamin D deficiency) and normal serum phosphorous levels (in chronic kidney disease). When the abnormal metabolites are corrected, the PTH comes back into the normal range. Long-standing secondary hyperparathyroidism however can result in autonomous parathyroid function, so-called tertiary hyperparathyroidism, which develops as the hyperplastic glands develop focal adenomatous regions. The distinction between secondary and tertiary hyperparathyroidism is usually made by the presence of hypercalcemia in patients with longstanding CKD. Those with tertiary hyperparathyroidism have hypercalcemia where as those with secondary hyperparathyroidism typically have hypocalcemia.
Familial hypocalciuric hypercalcemia (FHH) is an inherited condition due to inactivating mutations in the CaSR. The mutated CaSR does not properly sense calcium concentrations, and, as a result, the parathyroid gland produces excess PTH, erroneously sensing low calcium levels. The high PTH results in an increase in calcium reabsorption at the DCT. The CaSR at the kidney (present at the TALH) is also insensitive to the high calcium levels, further increasing calcium reabsorption, resulting in a very low urinary rate of calcium excretion (FECa <1%). These patients have life-long, moderate elevations in serum calcium, and this is generally a benign condition. The distinction between primary hyperparathyroidism and FHH is through 24-hour urine collection to measure calcium excretion, which is high in primary hyperparathyroidism, but low in FHH.
Hypercalcemia of malignancy: Malignancy can lead to hypercalcemia by three main mechanisms. One is a paraneoplastic syndrome related to production of a hormone known as PTH-related protein (PTHrp) also known as humoral hypercalcemia of malignancy. This is the most common cause of hypercalcemia of malignancy, accounting for approximately 80% of cases. PTHrP is a much larger peptide hormone than PTH but has enough structural similarity to interact with the PTH receptor (PTH-1R) and cause similar bone and kidney effects that lead to hypercalcemia. Paraneoplastic hypercalcemia due to PTHrP has been associated with several tumors, most commonly squamous cell tumors of the lung and oropharynx and renal adenocarcinomas. PTHrP is widely expressed during fetal development but in adults is expressed only by cells in the breast during lactation – a key mechanism to mobilize calcium from the bone into breast milk. Patients with increased production of PTHrP will present with high serum calcium, low serum PTH, but high PTHrP levels.
Several causes of hypercalcemia associated with malignancy are the different effects of the tumor on the bone. Tumoral cells can release systemic or local cytokines, which activate osteoclasts and increase bone breakdown, leading to high calcium with appropriate suppression of PTH.
Finally, in the case of lymphomas, extra-renal activity of 1a-hydroxylase leads to excess 1,25-(OH)2-vitamin D and increased calcium absorption from the gut which leads to hypercalcemia. PTH is also low in these cases.
Milk-alkali syndrome develops when large amounts of calcium carbonate supplements (ex: tums) are ingested that simultaneously cause increased calcium absorption and decreased renal calcium excretion due to the alkalosis. Milk-alkali syndrome (also called calcium-alkalai syndrome) can also be seen in patients with chronic kidney disease who are taking calcium containing phosphorus binders. Doses of > 4g of calcium per day with an absorbable alkali are typically required to lead to milk alkali syndrome. The hypercalcemia decreases glomerular filtration rate (GFR) and causes diuresis. Among other effects, hypercalcemia causes vasoconstriction and inhibition of the Na/K/2Cl channels in the nephron ascending loop of Henle, thereby decreasing water reabsorption in the descending loop of Henle. This results in hypovolemia and the consequent decrease in the GFR. The volume depletion leads to alkalosis and the clinical triad of hypercalcemia, alkalosis, and volume depletion occurs.
The incidence of milk-alkali syndrome is increasing due to misguided excess intake of calcium and vitamin D for “bone health”. These patients will have elevated calcium, elevated vitamin D with low PTH. The diagnosis is confirmed with a thorough history including use of any dietary supplements.
Hypervitaminosis D (vitamin D excess) causes hypercalcemia. When Vitamin D is very high, this facilitates excessive calcium absorption from the gut which will lead to hypercalcemia. PTH levels in this condition will be appropriately suppressed (low).
Other causes: Certain medications can lead to increased calcium levels, particularly the thiazide diuretics which increase renal calcium reabsorption. You can also see hypercalcemia with granulomatous disease (due to increased 1-α hydroxylase activity), and conditions that increase bone turnover such as thyrotoxicosis or immobility.
Hypocalcemia
Patients with hypocalcemia are generally more symptomatic than those with hypercalcemia, although the symptoms and signs of hypocalcemia also are related to the rate of change in calcium levels as much as the severity of hypocalcemia. Hypocalcemia leads to neuromuscular irritability, which can be manifested on physical exam through demonstration of the Chvostek sign (contraction of the facial muscles when tapping on the facial nerve) or Trousseau sign (carpal spasm with occlusion of the brachial artery with a BP cuff). This NEJM video shows an excellent example of these exam findings: https://www.youtube.com/watch?v=kvmwsTU0InQ.
The most common reason for a low serum calcium level is a falsely low measurement due to low serum protein levels. Because most serum calcium is protein bound, any condition leading to lower serum protein levels will result in a low measured serum calcium, while the ionized calcium remains stable. This scenario results in no clinical manifestations of hypocalcemia. Serum calcium can be adjusted to compensate for low serum proteins using the following formula: Adjusted serum Ca2+ = measured total serum Ca2+ + 0.8(4 – current serum albumin).
Hypoparathyroidism is the most common cause of low serum ionized calcium. Hypoparathyroidism may result as a complication of thyroid or parathyroid surgery, from autoimmune destruction, or due to congenital abnormalities of the 3rd and 4th pharyngeal pouches (e.g. DiGeorge Syndrome).
Some non-hypoparathyroid states (such as vitamin D deficiency, malabsorption) also can cause hypocalcemia if compensatory PTH secretion is inadequate to maintain normal serum calcium levels (Table 2). Bisphosphonate use in the setting of vitamin D deficiency is a common cause of hypocalcemia. Bisphosphonates decrease calcium release from bone, and without adequate increase in dietary calcium intake, serum calcium levels will decrease.
|
PTH mediated (low PTH) |
Non-PTH mediated (high PTH) |
|
Autoimmune disease Congenital hypoparathyroidism Surgery Radiation |
Malabsorption Dietary calcium/vitamin D deficiency Bisphosphonates PTH resistance |
Pseudohypoparathyroidism is a rare cause of low calcium due to PTH resistance. These patients have short stature and a distinctive shortening of the 4th and 5th digits, a phenotype known as Albright’s Hereditary Osteodystrophy. They have a mutation in the signaling of the G-protein coupled receptor, which prevents activation of adenylate cyclase and generation of the 2nd messenger cAMP in response to PTH binding to its receptor. Patients have low calcium, and high phosphorus as seen with primary hypoparathyroidism but high PTH levels. They often have other hormonal abnormalities as well, since several endocrine hormones signal via similar G-protein coupled receptors.
Treatment of Hypercalcemia and Hypocalcemia
Hypercalcemia: The treatment of hypercalcemia depends on the underlying cause. If the calcium level is severely elevated and/or symptoms are severe, urgent treatment is often needed. The immediate treatment for symptomatic hypercalcemia is IV saline infusion to increase glomerular filtration and increase passive calcium excretion. Loop diuretics (furosemide) can be added when adequate urine flow is established. Calcitonin (SQ or IM) works fairly rapidly (hours) but the effect lasts only 36-72 hours due to tachyphylaxis. Bisphosphonates (IV) decrease calcium slowly (over days), but the effect can last for weeks or even months, and these drugs have become the mainstay of treatment. Denusomab is also effective in treating hypercalcemia, especially in people with renal impairment. Bisphosphonates and denusomab are medications for osteoporosis and are discussed below.
Primary hyperparathyroidism is usually managed by surgical removal of the offending parathyroid adenoma(s). This is often assisted via pre-surgical localization procedures such as neck ultrasound or a nuclear medicine study (sestamibi scan) to localize the adenoma. Medical management with a CaSR agonist (cinacalcet) is available for those with primary hyperparathyroidism in whom surgery is contra-indicated or refused. Cinacalcet is also used to control PTH levels in patients with secondary hyperparathyroidism due to CKD. This medication activates the CaSR, acting as a calcium mimetic, which downregulates PTH release. Calcium levels must be followed closely for initial dose adjustment to avoid causing hypocalcemia.
For non-PTH mediated causes of hypercalcemia (malignancy, milk-alkali syndrome, etc.), the treatment is aimed at correcting the underlying cause.
Hypocalcemia: Therapy depends on the severity of the hypocalcemia, symptoms, rapidity of onset of hypocalcemia, and co-morbid conditions. The mainstay of treatment is oral calcium replacement, but IV calcium may be required for severe cases. In chronic hypocalcemia, vitamin D and thiazide diuretics are often used to help control calcium levels as well. In cases of primary hypoparathyroidism (PTH deficiency leading to hypocalcemia) patients require treatment with calcitriol (1,25-(OH)2 vitamin D) AND oral calcium. The calcitriol is required to ensure the oral calcium is absorbed from the gut because PTH is required to activate vitamin D from 25-OH Vitamin D to 1,25-(OH)2 vitamin D. Thus, in the absence of PTH, synthetic 1,25-(OH)2 vitamin D must be given for calcium to be absorbed.
Bone biology
An important element in the maintenance of normal calcium levels is the skeletal system. Bone is the main storage depot of calcium in the body, and mobilization of calcium from bone allows for the precise control of calcium levels that we normally see.
Structure
The extracellular component of bone consists of an organic matrix known as osteoid, primarily composed of type I collagen, and a mineral component, primarily composed of calcium and phosphate organized in a crystal structure termed hydroxyapatite. Mineralization of bone occurs along the osteoid “blueprint” within collagen fibrils. This relationship allows the skeleton to be lightweight but strong enough to withstand significant mechanical stress. Even small changes in the organization of the collagen fibrils can lead to a significant decrease in bone strength.
The cellular component of bone consists of the osteoblast, the osteocyte and the osteoclast. The primary function of the osteoblast is bone formation via osteoid production and promotion of mineralization (osteoBlasts Build bone). Osteoblasts are usually found on the surface of newly formed bone. The osteoblast contains cell surface receptors for most of the major hormones involved in bone metabolism, such as parathyroid hormone, vitamin D, and the sex steroids. During bone formation the osteoblast secretes proteins such as alkaline phosphatase and osteocalcin that promote bone collagen production and mineralization. These proteins can be measured in serum as a clinical marker of bone formation.
After the osteoid is formed and mineralized the osteoblast gets trapped in the mineral complex and becomes an osteocyte, the major cell type in bone. Osteocytes are mechanosensors that detect areas of mechanical stress and communicate this to surface osteoblasts in a paracrine fashion.
The osteoclast is predominantly responsible for bone resorption (osteoCLasts CLear out bone). Originating from a hematopoietic stem cell, the osteoclast undergoes maturation and differentiation from a precursor cell under the direct control of the osteoblast. A mature osteoclast can form a tight anchor to mineralized bone and secrete a highly acidic solution from its basolateral membrane that promotes mineral resorption.
Bone remodeling
Bone is a dynamic organ, and the skeleton is continually being remodeled in order to maintain its integrity. At any given time, approximately 10% of our skeleton is undergoing remodeling, with the entire skeleton “turning over” every decade. Remodeling is necessary to repair fractures and buffer weaker areas of the skeleton exposed to mechanical stress. It will also allow for mobilization of calcium from bone in times of need. The process is tightly orchestrated and occurs in a sequential fashion of osteoclastic bone resorption followed by osteoblastic bone formation, in a complex known as the bone multicellular unit (Figure 4). During remodeling, bone resorption occurs more efficiently than bone formation. For example, resorption of a 10-micron area of bone occurs in three weeks while the same area takes three months to rebuild. Any process that increases bone resorption (hyperthyroidism, hyperparathyroidism, hypogonadism, aging), will therefore result in bone loss.
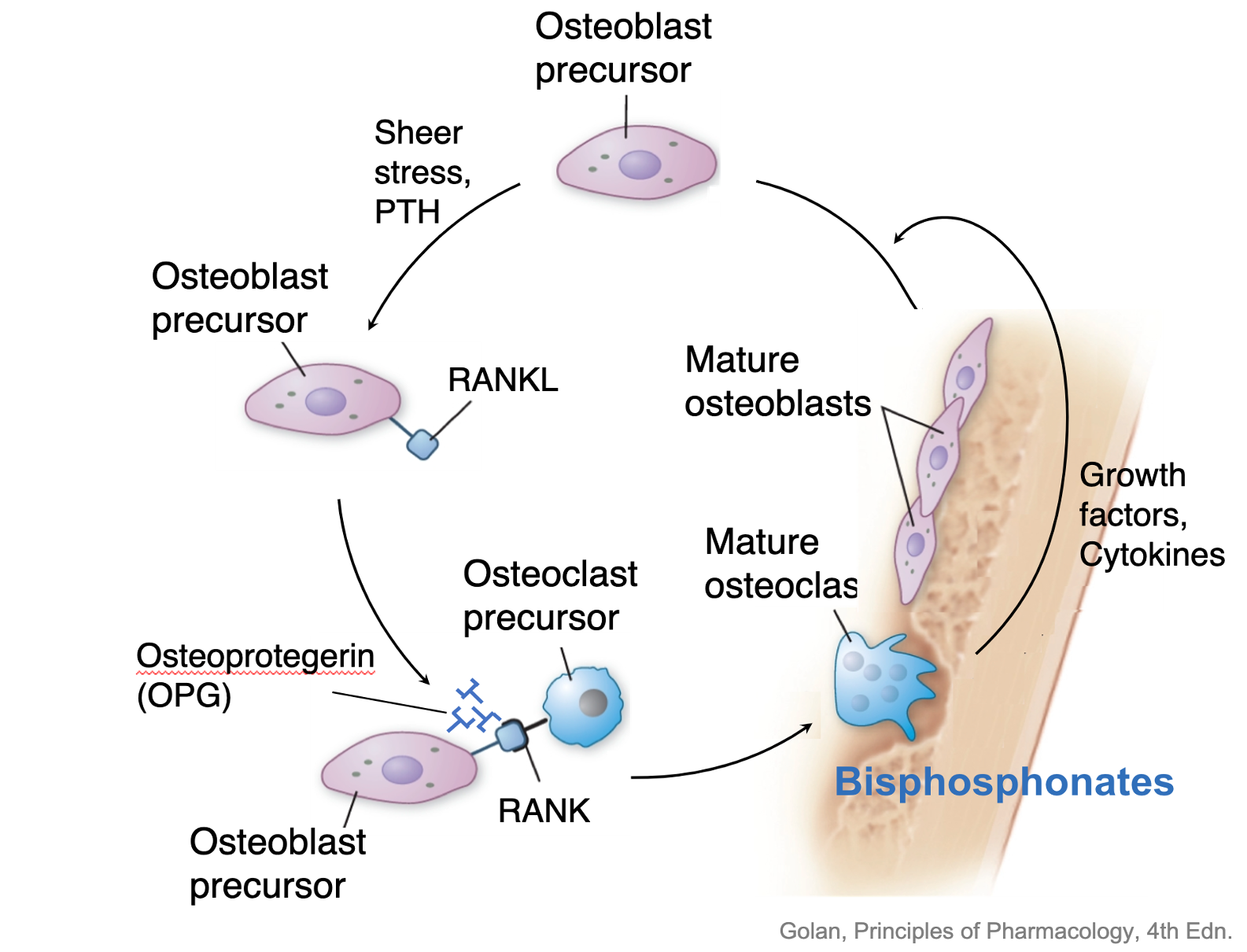
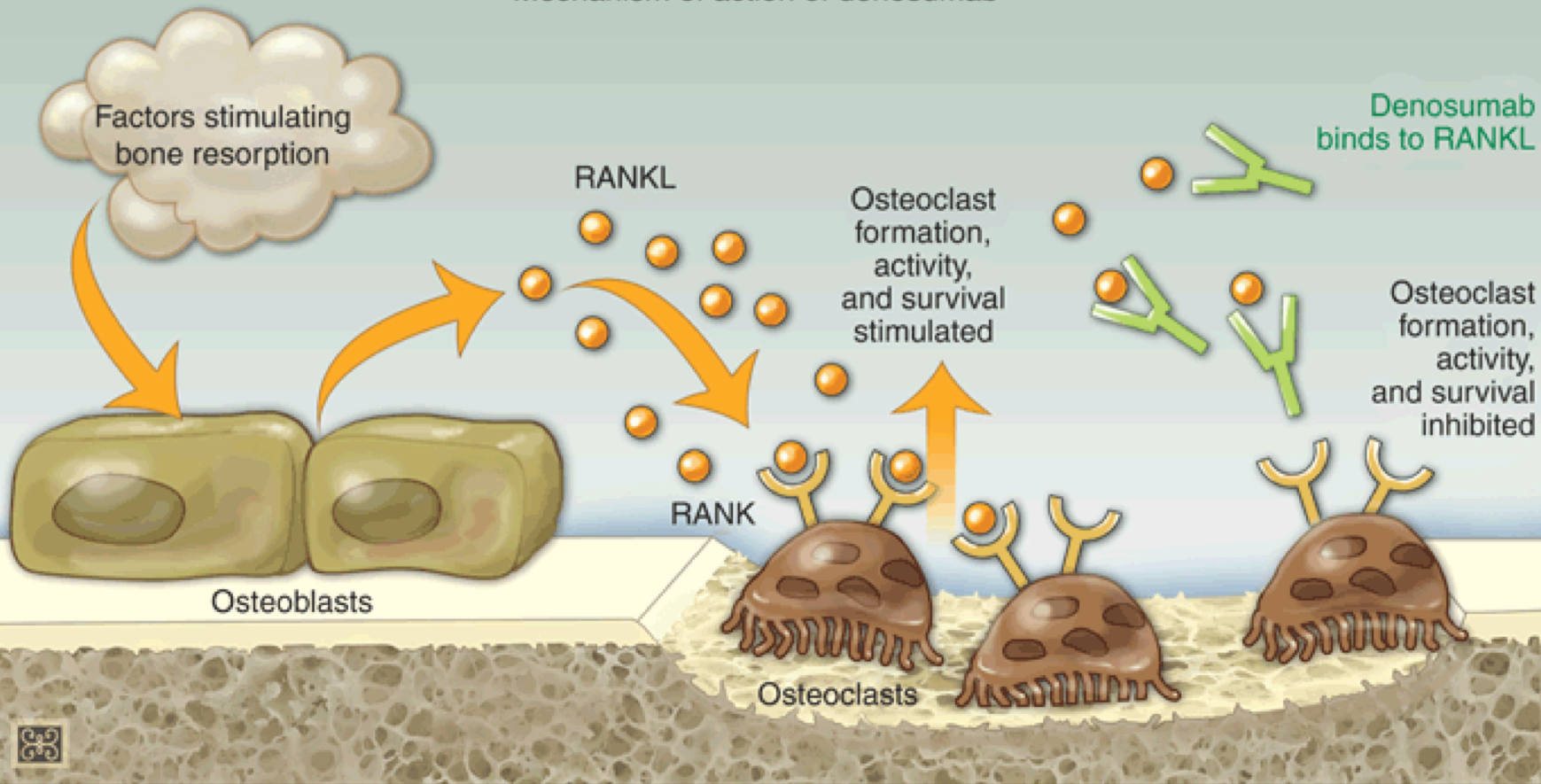
The cellular signals contributing to the coordination of bone remodeling have been identified. The osteoblast secretes RANKL (receptor activator of nuclear factor kappa B ligand), a member of the tumor necrosis family of receptors. RANKL binds to RANK on the surface of pre-osteoclasts to stimulate osteoclast maturation and activity. Osteoblasts also secrete a soluble protein called osteoprotegerin (OPG), which acts as a decoy receptor, binding to RANKL and inhibiting RANKL-RANK interaction. This inhibits osteoclast recruitment and action (Figure 5). The interplay between RANKL-RANK-OPG will determine whether bone resorption or bone formation predominates. When RANKL secretion exceeds OPG, osteoclasts are activated leading to bone resorption. When OPG is the predominant product, there is decreased osteoclast activation, and bone resorption is inhibited.
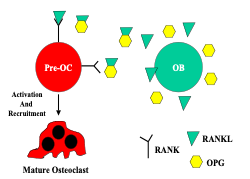
Another protein involved in bone remodeling, sclerostin, is produced by the osteocytes. Sclerostin inhibits osteoblast differentiation, and stimulates RANKL production, which increases bone resorption. Individuals with a loss of function mutation in the sclerostin gene develop a rare bone disorder called sclerosteosis. This disorder is characterized by high bone mass and increased bone mineral density (BMD) due to unopposed bone formation.
The balance between formation and resorption is coordinated and changes throughout life. During skeletal growth, bone formation is greater than bone resorption and bone mass accrues. Bone density reaches a peak in early adulthood (age 25-30) and then enters a relatively quiescent phase until about age 40. In subsequent years, bone resorption exceeds formation, a process known as age-related bone loss. The bone density decline associated with aging varies per individual and by skeletal site but averages about 0.5%/year.
Many of the hormones involved in bone metabolism function by altering the RANK-RANKL-OPG balance to promote either bone formation (estrogen) or bone resorption (PTH, vitamin D). Bone resorption will mobilize skeletal calcium stores and is a valuable process to avoid hypocalcemia.
Regulation of bone
Estrogen
Both estrogen and testosterone are important for proper skeletal development and integrity. Hypogonadism (low estrogen or low testosterone) in all patients is associated with a decrease in peak bone density, an increase in age-related bone loss, and an increase in fracture rates. Estrogen increases osteoblastic production of osteoprotegerin (OPG), inhibiting osteoclast function (and thus inhibiting bone resorption). This improves the bone formation to bone resorption ratio. Testosterone is aromatized to estrogen and is critical for maintaining normal bone mass in patients where testosterone is the primary sex hormone.
Since sex hormones play a key role in maintaining bone mass, there are significant differences in risk of bone loss among different sexes. During the perimenopausal period the loss of estrogen increases the rate of bone loss dramatically, with some females losing 2-5%/year. Because males achieve a higher peak bone mass and do not incur the significant bone loss associated with menopause, they tend to reach fracture thresholds five to ten years later than females.
Trans-women are at higher risk of osteoporosis if they are using high dose androgen blockers or have undergone gonadectomy and are not taking adequate estrogen replacement. Trans-men have higher risk of osteoporosis if they have had oophorectomy prior to age 45 without optimal testosterone replacement. For other populations, the risk of osteoporosis is increased with any interventions or pathology that result in low levels of sex hormones. Any patient who has low sex hormones due to surgery or medical therapy would also be at higher risk of osteoporosis.
Parathyroid hormone
PTH is another hormone that plays a key role in regulation of bone resorption. PTH binds to receptors on the osteoblasts, ultimately leading to an increase in RANKL produced by the osteoblast. This results in stimulation of osteoclasts to increase bone resorption, leading to increased serum levels of calcium and phosphorous. PTH also induces IL-1 expression in osteoblasts which leads to osteoclast activation. While this process is normally tightly regulated, with higher calcium levels suppressing PTH and stopping bone resorption, any process that results in abnormally elevated PTH levels will lead to excessive bone resorption.
Vitamin D
The skeleton, like all vital structures, is dependent on appropriate nutritional support for survival and optimal function. Delivery of appropriate amounts of protein, vitamins and minerals is integral to maintenance of skeletal health. Calcium, phosphorus, and vitamin D are all key factors in normal bone mineralization.
The impact of vitamin D deficiency on the skeleton is dependent on the degree of insufficiency/deficiency and the age of the patient. Vitamin D deficiency causes a decrease in bone mineralization, poor bone growth and secondary hyperparathyroidism. Severe deficiency leads to excess osteoid (non-mineralized collagen matrix) content of bone termed rickets in childhood and osteomalacia in the adult. These disorders will be discussed in detail in the section below. Milder forms of vitamin D deficiency will lead to compensatory parathyroid-induced calcium resorption from bone. This can result in a decrease in peak bone mass achievement and an increased rate of bone loss later in life.
Mechanical loading
Mechanical strain is beneficial to the skeleton if it does not exceed the fracture threshold. Osteocytes are the most abundant cell in the skeleton and secrete the hormone sclerostin, an antagonist of osteoblast activation. They also have mechanoreceptors that can detect mechanical strain on the bone. In the presence of sclerostin, osteoblast activity is suppressed. Mechanical strain reduces osteocyte production of sclerostin thereby promoting osteoblastic activity in this area, presumably to ensure bone formation at sites of stress. Clinically this phenomenon manifests as a higher BMD among physically active individuals and partially explains the direct correlation of BMD with weight. Epidemiologic studies reveal that individuals with obesity are less likely to fracture and leaner individuals more likely to do so. Conversely, patients with spinal cord injuries can lose up to 30% of their BMD during the first year following the accident, due to loss of weight bearing.
Bone Pathology
Osteoporosis
Osteoporosis, as defined by the National Osteoporosis Foundation, is a systemic skeletal disorder characterized by compromised bone strength predisposing to an increased risk of fracture. Osteoporotic bone is histologically similar to normal bone, but less of it exists due to losses from different etiologies. Osteoporotic fractures can occur at any skeletal site, but vertebral fractures are the most prevalent, and hip fractures associated with the highest morbidity and mortality.
Osteoporosis can be diagnosed clinically in any individual suffering a spontaneous fracture or a fracture after minimal trauma (impact equal to a fall from standing height), termed a “fragility fracture.” Measurement of bone mineral density can diagnose osteoporosis in the absence of a clinical fracture. Dual energy X-ray absorptiometry (DEXA) uses minimal radiation to non-invasively measure bone mineral density (BMD). The patient’s BMD is compared to the average BMD of a younger sex-matched population at peak bone mass (age 20-30 years). Each BMD standard deviation from the mean of the reference population equals one T-score equivalent, with positive T-scores above the average and negative T-scores below. For example, an elderly female with a BMD score 2 standard deviations below the mean BMD of a young female population would have a T-score of –2.0. Osteoporosis is defined as a T-score of negative 2.5 or less. Multiple epidemiological studies have confirmed the association between BMD and fracture risk, but have found the risk differs for different populations, as both sex and race influence the risk of fracture.
Current guidelines for diagnosis recommend using a non-race adjusted database for diagnosing osteoporosis. This is controversial. Several observational studies have found that fracture risk differs for different ethnic groups, suggesting a need for different normative databases for each ethnicity. However, defining ethnicity is difficult. It is a complex interaction between genetics, geography, culture and society. It is impossible to define clinically. As well, at least one study comparing fracture rate in various ethnic groups using a white reference population, found similar fracture risks for each SD reduction in BMD regardless of patients’ self-identified race. Thus, the International Society for Clinical Densitometry (ICSD) recommends using a standard database for all patients.
With the 2019 guidelines, the ICSD provided specific guidelines for evaluation of bone density in transgender and gender non-conforming individuals. For individuals on gender confirming hormone therapy, there is no increased risk of osteoporosis compared to the cisgender population. If individuals are without hormone therapy for >1 year, they are at risk of bone loss. For purposes of T-score calculation, the database that matches the individual’s gender identity should be used, as there is no difference in fracture risk between cisgender and transgender individuals. For gender non-conforming individuals, current guidelines recommend using the database that matches the individuals assigned sex at birth.
Multiple factors may contribute to the development of osteoporosis (Table 3). The most important of these: age and genetic predisposition, are non-modifiable. It is imperative for the clinician to search for contributing factors that may, if identified and treated, reduce a patient’s future fracture risk. Usually this can be accomplished with a complete history and physical and a tailored laboratory evaluation for secondary causes of osteoporosis.
|
Non-modifiable |
Medical conditions |
Medications |
Nutrition and Lifestyle |
|
Age |
Renal disease |
Steroids |
Low vitamin D |
|
Genetics |
Liver disease |
Anti-epileptics |
Low calcium |
|
|
Hyperthyroidism |
Thiazolidinediones |
Alcohol use |
|
|
Hyperparathyroidism |
|
Tobacco |
|
|
Hypogonadism |
|
Caffeine |
|
|
Malabsorption |
|
Immobility |
|
|
Chronic inflammation |
|
Low body weight |
There are substantial disparities in screening for and treatment of osteoporosis. Screening for osteoporosis is low for all populations. 33% of Caucasian women receive appropriate screening, 5-15% of African American populations receive screening. Medicare covers DEXA screening for all women over age 65. Treatment disparities for osteoporosis also exist, with fewer non-Caucasians receiving treatment for osteoporosis at all levels of fracture risk. While hip fracture is less common in non-Caucasian individuals, the outcomes of hip fracture are worse, so it is critical that all patients are aggressively screened for osteoporosis risk factors, get appropriate DEXA testing, and receive treatment when indicated.
Treatment of osteoporosis
Multiple pharmacologic agents are available to reduce the risk of fracture for patients with osteoporosis. Mechanisms of action and side effects will be covered in the associated Bone/Osteoporosis Pharmacology lecture. Most pharmacologic agents for osteoporosis act by reducing bone resorption.
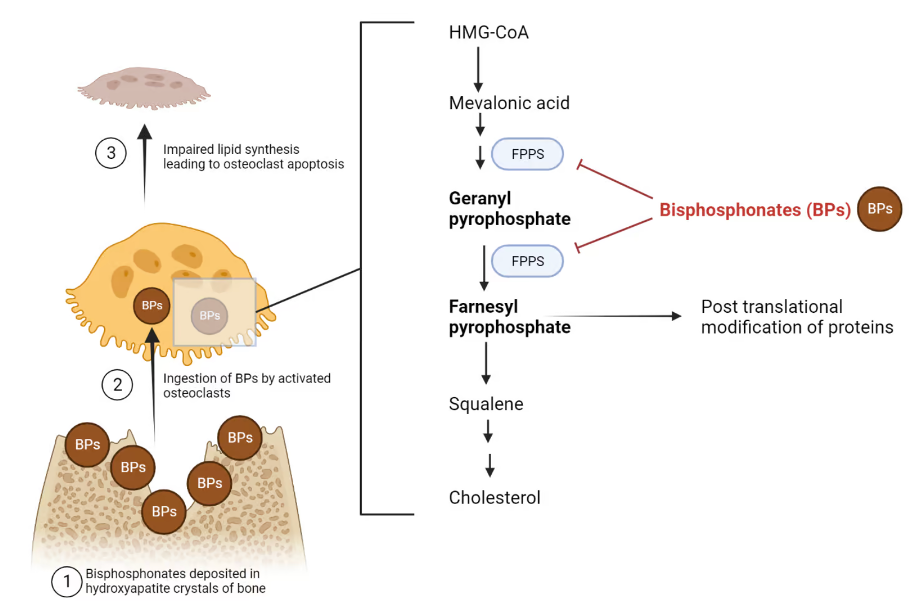
The most commonly used anti-resorptive agents are the bisphosphonates (Fig 6). These can be given orally (daily or weekly) or as IV infusions (quarterly or annually). They act by inhibiting osteoclast-mediated bone resorption, which slows bone loss. There is very strong data to demonstrate fracture prevention with use of these medications. The main side effect with the oral bisphosphonates is esophagitis. There have been rare cases of osteonecrosis of the jaw, particularly with the IV forms when given in high doses. Another rare side effect is atypical femur fracture, which occurs when these medications are used for longer than the recommended duration (3-10 years depending on the formulation and severity of the osteoporosis). The bisphosphonates are renally excreted, so they cannot be used in patients with kidney disease. These medications are considered first line treatment for most people with osteoporosis.
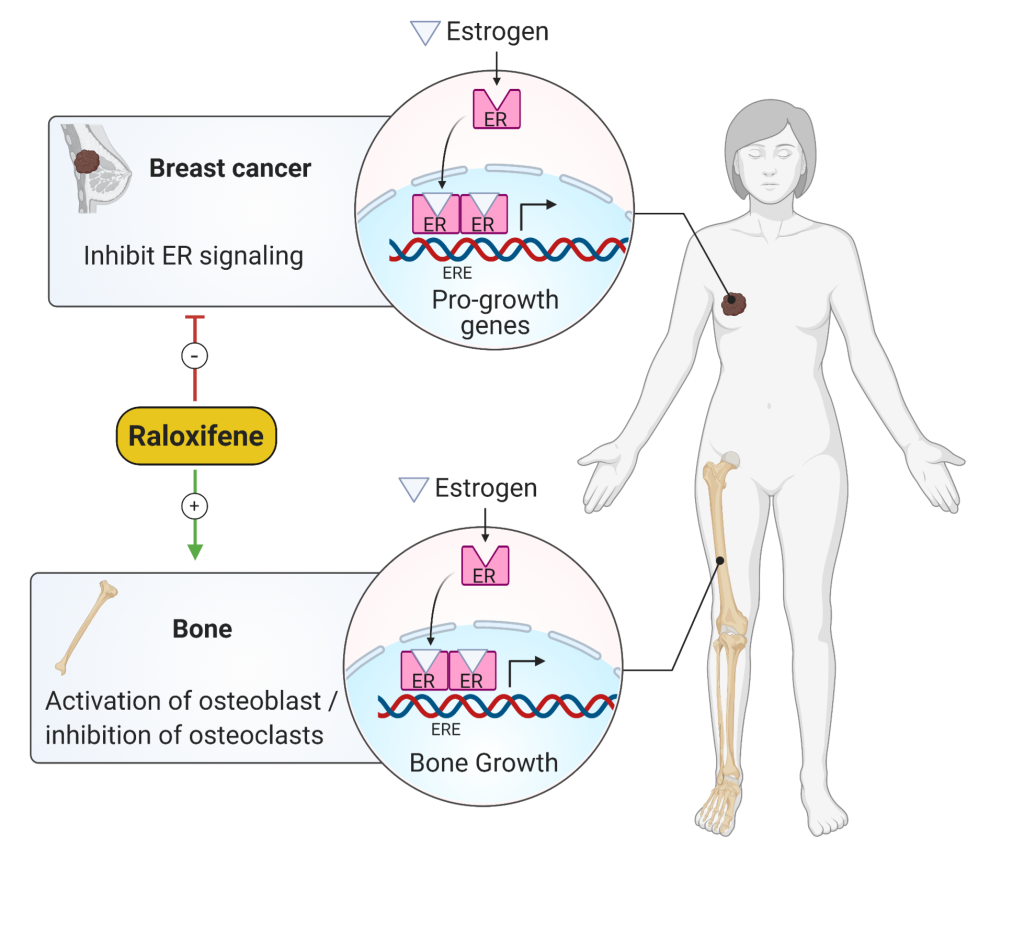
Selective estrogen receptor modulators (SERMs) (Fig 7), such as raloxifene, are another option for treating osteoporosis. These drugs act at the estrogen receptor but have different effects in different tissues. Raloxifene has positive effects in the bone, slowing bone loss, but antiestrogenic effects in the uterus, so unlike tamoxifen, another SERM, it will not increase the risk of endometrial cancer. It also has antiestrogen effects at the breast, so is a good option for post-menopausal patients at high risk of breast cancer, who need treatment for osteoporosis. It does cause hot flashes and can increase the risk of venous thromboembolisms.
Denosumab (Fig 8) is a newer anti-resorptive agent for the treatment of osteoporosis. This is a monoclonal antibody which inhibits RANKL. You will recall from a previous section that the osteoblast secretes RANKL which stimulates osteoclast maturation and activity. Denosumab prevents this interaction, and blocks osteoclast mediated bone resorption. This medication is given subcutaneously once every 6 months. It is not renally cleared, so is one of the only osteoporosis medications that can be used for patients with CKD. Its use is limited by cost – one injection can cost upwards of $1,000. It can cause hypocalcemia, so should not be given to patients with hypocalcemia at baseline, and calcium levels should be monitored during therapy.
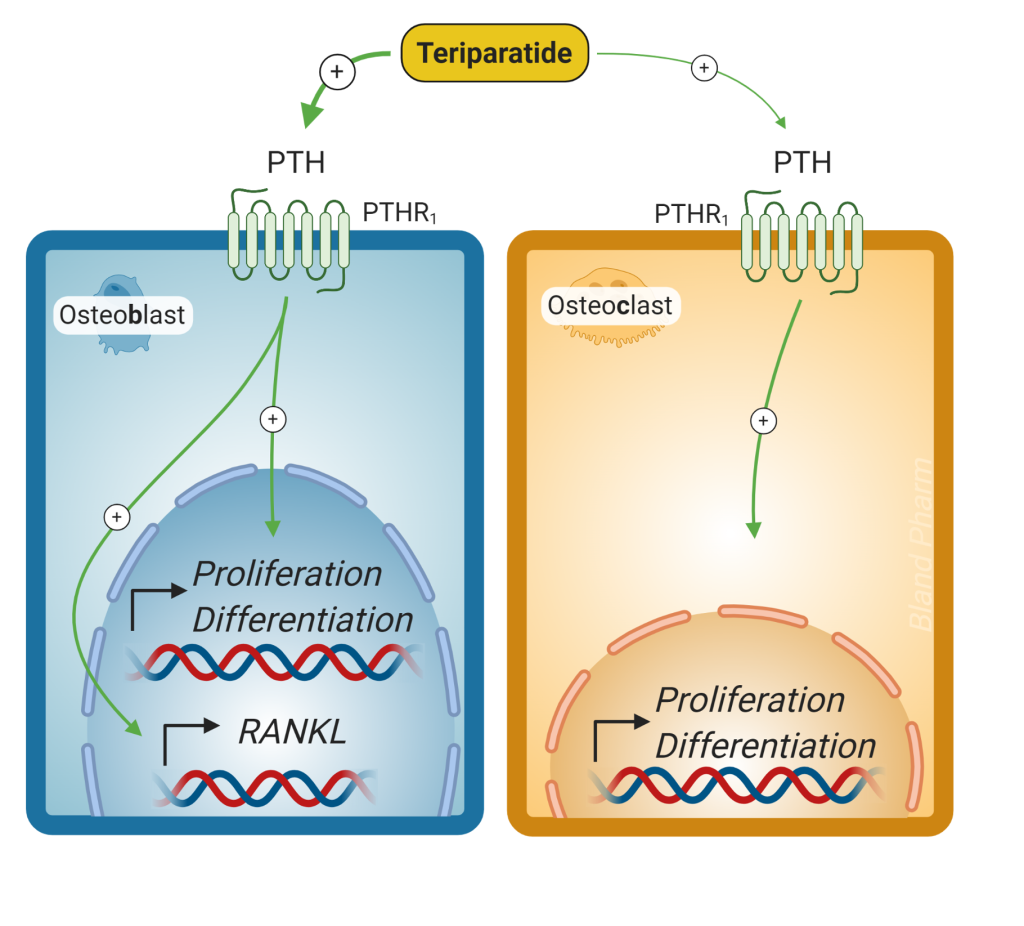
For patients with severe osteoporosis, preventing further bone loss may not be enough. Medications are available for severe osteoporosis that stimulate bone resorption. These are PTH analogs (Fig 9) that are given as a daily subcutaneous injection. Chronic exposure to high PTH levels, as seen with hyperparathyroidism, can cause bone loss. However, intermittent exposure preferentially activates osteoblasts resulting in an increase in bone formation. Teriparatide is typically only used for 2 years of use, but in certain cases (for individuals with very high fracture risk) it can be used for longer periods of time. Because it causes bone growth, it should not be used in people who are at increased baseline risk of osteosarcoma (those with prior bone radiation, history of osteosarcoma and Pagets disease of the bone). It should be followed with an anti-resorptive agent (bisphosphonate or denosumab) to prevent further bone loss.
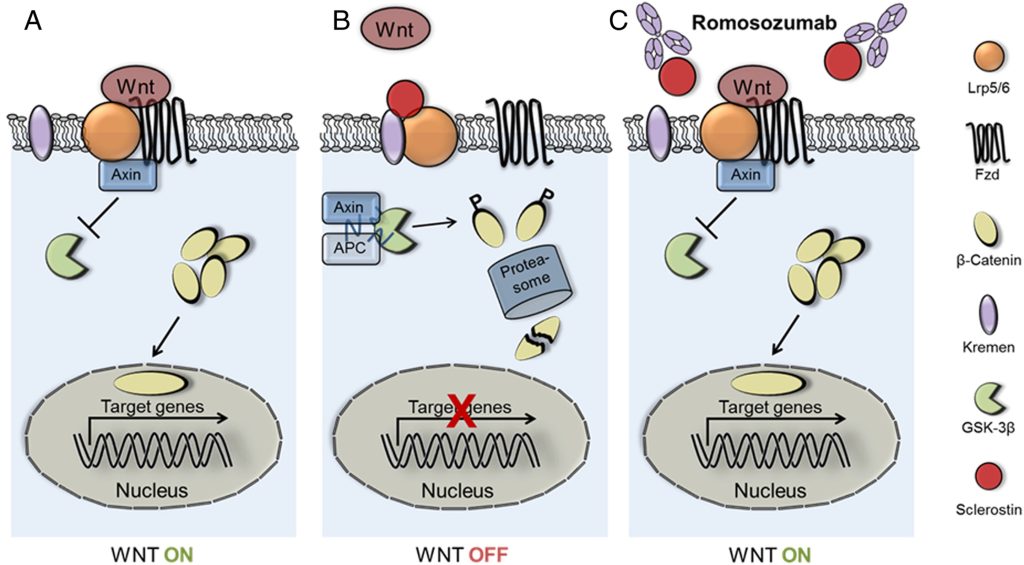
The newest medication for the treatment of osteoporosis is romosuzamab (Fig 10). This medication is an anti-sclerostin antibody and leads to increased bone formation and decreased fracture risk. Romosuzamab is given as a once monthly injection for 12 months. The main side effect is injection site reactions. One trial did show an increased risk of CV events, so this should not be given to patients with a history of or at high risk for CVD.
Glucocorticoid-induced osteoporosis
Glucocorticoids (both endogenous and exogenous) can lead to low bone mineral density when in excess. Even at low concentrations, glucocorticoids cause osteoblast apoptosis. They promote calciuresis with a compensatory rise in PTH. They also inhibit calcium absorption from the gut. Long-term supraphysiologic corticosteroid use results in hypogonadism and suppression of growth hormone, effects that are all deleterious to skeletal health.
Epidemiologic studies have confirmed the association between glucocorticoid use and fracture. Any skeletal site can be affected, but vertebral fractures are most common. Fractures can occur at any age and are dose-dependent. Both bisphosphonates and teriparatide have been shown to prevent glucocorticoid-induced bone loss and to reduce fracture incidence. Either of these therapies should be considered in individuals requiring supraphysiologic doses of glucocorticoids for greater than three months or with a history of Cushing’s syndrome.
Osteonecrosis
Also known as aseptic necrosis or avascular necrosis (AVN), osteonecrosis is a pathologic process that has multiple etiologies. Etiologies include glucocorticoid excess (both exogenous or endogenous), sickle cell disease, alcoholic use disorder, and idiopathic (Legg-Calvé-Perthes disease). Essentially, this condition is caused by infarction of the bone and the bone marrow. This lack of blood flow leads to death of the bone and mechanical failure. The process is often very painful and progressive. The most affected sites include the distal femur, the humeral head, and the small bones of the wrist and foot. Osteonecrosis of the jaw is a known, very rare side effect of bisphosphonate medications.
Osteomalacia
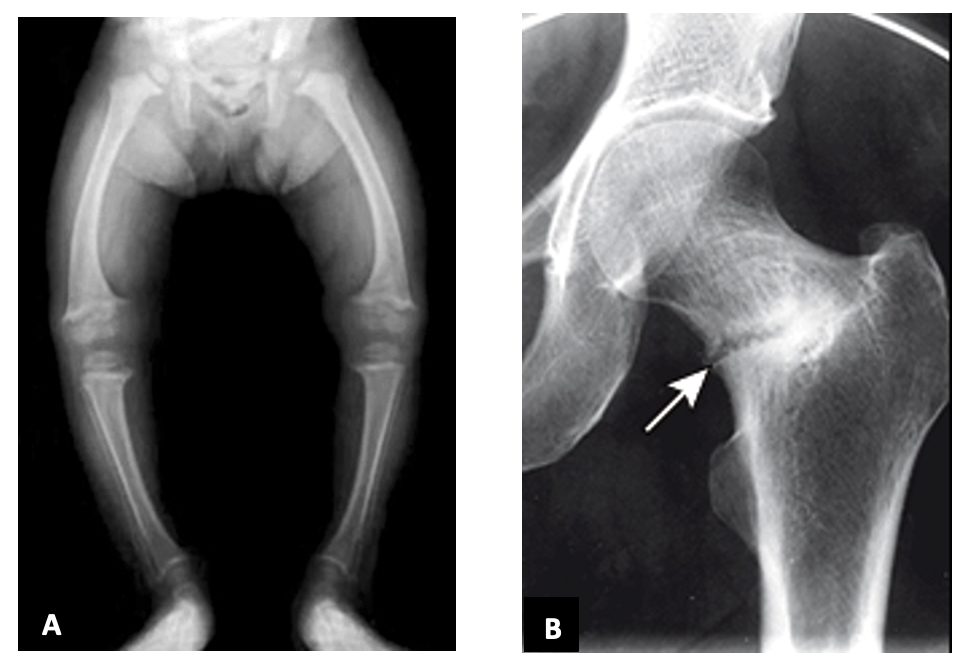
Osteomalacia is a skeletal condition resulting from an inability to adequately mineralize the collagen matrix during bone formation. Histologically this bone has increased osteoid (non-mineralized collagen) content. Osteomalacic bone is characterized by high pliancy and decreased strength. If the condition occurs prior to fusion of the growth plates (in childhood) it is termed “rickets”. Rickets in children is usually due to nutritional deficiency of vitamin D, or in rare cases abnormalities in vitamin D metabolism, such as defects in 1α-hydroxylase activation, or congenital vitamin D receptor abnormalities. Children with rickets have stunted growth and bowing of the long bones (figure 11a) with multiple fractures and joint malalignment, leading to pain. On imaging, they have widening of the epiphyseal plate and Looser zones (figure 11b), which represent pseudo fractures.
In adulthood the condition is known as osteomalacia. Clinical manifestations include fractures, bone pain, and decreased muscle strength. Osteomalacia is usually caused by inappropriate ingestion, absorption, or metabolism of vitamin D. Severe vitamin D deficiency, either from inadequate dietary intake, or fat malabsorption will lead to osteomalacia if it goes untreated for a prolonged period of time. Phosphate is also necessary for proper bone mineralization, and disorders resulting in low phosphate can lead to osteomalacic bone. Patients with osteomalacia present with bone pain, fracture or difficulty with walking. Treatment of osteomalacia depends on correction of the underlying etiology and repletion of vitamin D and/or phosphate.
Paget Disease of the Bone (aka osteitis deformans)
Paget Disease of the bone will be covered in another block, but for completeness is detailed here as well. You will not be tested on Paget Disease during the CHB block. Paget Disease of the Bone (aka osteitis deformans) is a chronic bone disorder that affects typical bone remodeling. There is an accelerated rate of bone remodeling (increased activity of both osteoclasts and osteoblasts), which leads to bone overgrowth at one or multiple sites. This disorder occurs in three phases that present in this order (1) osteolytic phase (osteoclasts dominate), (2) mixed osteolytic-osteoblastic stage, (3) osteosclerotic phase. It begins in late adulthood and increases with age. An estimated 1% of the US population is affected. This condition is more common in the United Kingdom, central Europe, and Greece and in areas colonized by European immigrants. Both genetic and environmental factors play a role in development of this condition. Paget disease can be monostotic (15% of cases, affecting only one site) and polyostotic (85% of cases, affecting multiple sites). Most cases are asymptomatic and discovered incidentally with x-ray or with alkaline phosphatase elevation. This enzyme is elevated due to the increased bone turnover. Calcium and parathyroid hormone are normal. Pain of the affected bone is the most common symptoms and may be due to microfractures or nerve compression from overgrown bone. The integrity of the affected bone is also abnormal, increasing risk for fracture at those sites. Due to bone overgrowth, there may be compressive symptoms that bring patients to clinical attention such as hearing loss or nerve impingement. More rare clinical features include enlargement of the craniofacial skeleton which can change the face shape and/or cause the head to be very heavy. Hat size may also change. Excessive weight bearing can cause bowing of the femurs and the tibias. Rarely, the bone has overgrown so significantly that an individual may develop high-output heart failure due to increased blood flow to the bone. People with Paget Disease of the Bone are at increased risk for osteosarcoma. Bisphosphonates are the treatment of choice because these medications slow or stop bone turnover, which addresses the underlying abnormality.