
CHB Chapter Uploads
5 Hormones of the adrenal medulla
Epinephrine is almost exclusively secreted by the adrenal medulla and constitutes about 85% of the catecholamines produced in the normal adrenal medulla. The two major sources of circulating norepinephrine are the postganglionic sympathetic nerve endings and, to a lesser extent, the adrenal medulla.
Catecholamines are synthesized from tyrosine which is converted via a series of steps to 1-3,4-dihydroxyphenylalanine (DOPA), dopamine, norepinephrine, and epinephrine. The first step is catalyzed by tyrosine hydroxylase (tyrosine DOPA), and it is the rate-limiting step in the pathway of catecholamine biosynthesis. The methylation of norepinephrine to form epinephrine requires the glucocorticoid-dependent enzyme phenylethanolamine-N-methyltransferase (PNMT), which is found primarily in the adrenal medulla. Therefore, the high concentrations of corticosteroid that are provided by the portal blood supply from the adrenal cortex to the medulla are important for normal production of epinephrine.
Catecholamines have a very short half-life in the circulation. Catecholamines are converted to metanephrine and normetanephrine, and then by monoamine oxidase (MAO) to vanillylmandelic acid (VMA). The catecholamine metabolites have a significantly longer half-life than the catecholamines, and these are the hormones typically measured when there are concerns for catecholamine excess.
Actions of catecholamines are mediated by three major classes of receptors (α1, α2 and ß) on the surface of target cells. Actions at the α1-receptors lead to vasoconstriction, increased cardiac contractility and sweating; the α2-receptors regulate norepinephrine release from sympathetic nerves, while actions at the beta receptor lead to increased heart rate. Together these actions lead to increased cardiac output and increased peripheral vascular resistance. Epinephrine is also a “counter-regulatory” hormone in glucose metabolism and its release is stimulated by hypoglycemia to induce hepatic glycogenolysis.
- Adrenal Nodules
Adrenal nodules (unilateral or bilateral) are discovered frequently on imaging studies (eg. CT scan) that are done for another purpose. These masses are called “adrenal incidentalomas”. Discovery of adrenal nodules raises three questions to determine how these masses should be treated and followed: (1) is it malignant, (2) is it a pheochromocytoma, (3) is it functioning (catecholamine, aldosterone, or cortisol secreting). These questions can be answered with laboratory hormonal evaluation, history, physical, and dedicated adrenal imaging. Laboratory evaluation will be covered in the sections specific to the hormone being secreted.
Malignancy is an uncommon cause of adrenal tumors in people without a known diagnosis of cancer. Adrenocortical carcinoma is a very rare cause of adrenal tumors. On the other hand, the adrenal glands are common sites of metastatic disease, especially lung, kidney, breast cancers and melanoma. The size of the tumor and the imaging phenotype can predict likelihood for malignancy. When the size is > 4cm, it is more likely to be adrenocortical carcinoma. Therefore, unilateral removal of all adrenal masses > 4cm is recommended to avoid missing adrenocortical carcinoma.
The imaging appearance can also help differentiate between cortical (originating from the cortex) adenoma, pheochromocytoma, or malignancy. When CT scans are dedicated to evaluating the adrenal glands, the density of the nodule (without contrast) and the percent washout of contrast over time are both useful features. When the nodules is less dense (lipid rich) it is highly likely it is benign. When nodules have a rapid washout of contrast, they are more likely to be benign. Malignant nodules (adrenocortical carcinoma and metastases) tend to be irregular in shape, calcified, large, denser, and have delayed washout of contrast. Pheochromocytomas tend to be highly vascular, have medium contrast washout, and have increased density.
In some cases, the adrenal tumor is biopsied to distinguish adrenal adenoma from metastatic disease. Hormonal evaluation should be completed prior to adrenal biopsy.
- Primary hyperaldosteronism
- The most common cause of secondary hypertension is increased production of aldosterone. Secondary hypertension (as opposed to primary or essential hypertension) is high blood pressure that is caused by another medical condition. It is likely an underdiagnosed cause of hypertension, responsible for up to 15% of cases of hypertension. The most common causes of hyperaldosteronism are bilateral hyperplasia or unilateral aldosterone producing adenoma (Conn’s syndrome).
- Pathophysiology and clinical features
- Aldosterone leads to an increase in renal sodium absorption. This increases water reabsorption, leading to increased blood volume and increased blood pressure. At the same time, sodium absorption is balanced by renal excretion of potassium, leading to hypokalemia with elevated urine potassium levels. Aldosterone excess also leads to excessive excretion of hydrogen ions. The common clinical features of hyperaldosteronism are hypertension, hypokalemia, and metabolic alkalosis.
- Diagnosis of hyperaldosteronism
- Since the only clinical feature of hyperaldosteronism for most people is hypertension, it is important to screen certain individuals for elevated aldosteronism. Identifying elevated aldosterone can help direct antihypertensive therapies. Current guidelines recommend screening patients with the following clinical pictures for elevated aldosterone:
- Hypertension (HTN) with hypokalemia
- Severe hypertension (SBP >150 mmHg or DBP >100 mmHg)
- HTN not well controlled on 3 medications
- HTN with an adrenal nodules
- HTN onset at a young age (< 40 years)
Screening for hyperaldosteronism includes measuring both aldosterone levels and renin activity, as aldosterone production is under the control of renin. If aldosterone is elevated with low renin, this suggests primary hyperaldosteronism. If both renin and aldosterone are elevated however, it suggests there is another cause of the patient’s hypertension.
Once the diagnosis of hyperaldosteronism is established, it is important to determine if the source is unilateral, which can be treated surgically, or bilateral, which can only be treated with medications. This is done with a procedure called adrenal vein sampling (AVS). In this procedure, a catheter is inserted into each adrenal vein, and the levels of aldosterone are measured and compared to the levels of aldosterone from peripheral blood. If both adrenal glands are producing excess aldosterone, the levels will be similar on both sides. If one gland is producing excess aldosterone, the levels on that side will be 4 x greater than the unaffected side.
- Treatment of hyperaldosteronism
Hyperaldosteronism due to a unilateral aldosterone producing adenoma can be treated with adrenalectomy (adrenal gland removal). This will often cure the hypokalemia and decrease the severity of hypertension. For patients who are not surgical candidates due to bilateral aldosterone production, other comorbidities or personal preference, medical treatment for hyperaldosteronism is with spironolactone. Spironolactone is a mineralocorticoid receptor blocker. This medication acts to block the effects of aldosterone, lowering blood pressure and improving hypokalemia. Due to some androgen blocking effects, spironolactone can lead to gynecomastia (breast growth) and hypogonadism.
- Glucocorticoid excess (Cushing‘s syndrome)
The term Cushing’s syndrome (Harvey Cushing was a pituitary surgeon) refers to the signs and symptoms of chronic glucocorticoid excess, whether from taking glucocorticoid medications (exogenous) or from producing too much cortisol (endogenous). Endogenous cortisol excess may come from an adrenal adenoma or carcinoma or may be caused by excess ACTH signaling, either from a pituitary adenoma (Cushing’s disease) or an ectopic (non-pituitary) source of ACTH.
Signs and symptoms of Cushing’s syndrome
Patients with Cushing’s syndrome can have many symptoms and signs caused by glucocorticoid, mineralocorticoid or androgen excess (table 2). The effects of glucocorticoids are summarized below.
Muscle and fat
The breakdown of muscle tissues causes muscle atrophy and weakness and increased muscle insulin resistance can cause hyperglycemia. Excess cortisol signaling in adipose tissue stimulates growth of fat cells and increases the deposition of fat in certain areas of the body – central fat, liver fat, fat on the face and trunk. In contrast, subcutaneous fat stores on the limbs undergo lipolysis and shrink. The mechanism for this differential effect on trunk vs. limb subcutaneous fat is not known.
Connective tissue and bone
Cortisol excess has negative effects on skin and bone. Cortisol mobilizes amino acids from collagen, skin and connective tissue, which can lead to thin skin, violaceous striae on the abdomen, and bruising (capillary fragility). Glucocorticoids also act at bone to inhibit formation (osteoblasts) and stimulate resorption (osteoclasts via an osteoblast signal). Glucocorticoid excess also inhibits synthesis of vitamin D, leading to decreased intestinal calcium absorption and increased urinary calcium excretion. This contributes to the bone loss and increased fractures that can occur with cortisol excess.
Cardiovascular
In states of excess cortisol secretion, the 11βHSD2 activity that normal inactivates cortisol becomes overwhelmed. This allows cortisol to act at the mineralocorticoid receptor causing hypertension and hypokalemia.
Growth
Glucocorticoid excess inhibits growth in children, both through a direct effect on bones, and through decreased growth hormone (GH) and insulin-like growth factors (IGFs).
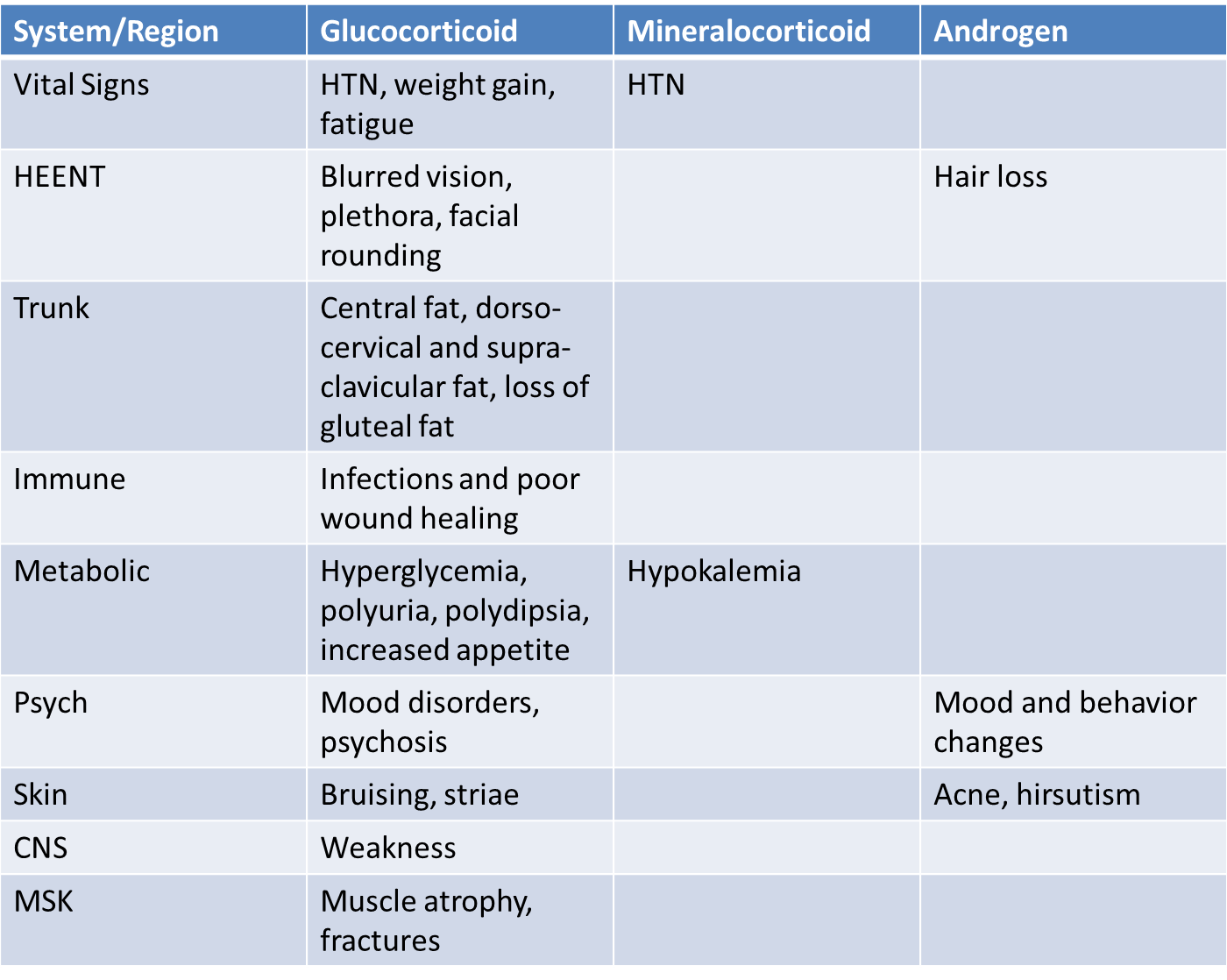
Table 2. Signs and symptoms of Cushing’s Syndrome
While many signs and symptoms of Cushing’s are non-specific, the most sensitive signs on physical exam for the diagnosis of Cushing’s syndrome include supraclavicular fat (which is an unusual finding even in patients with obesity), wide and purplish-red striae (rather than narrow and pale) and proximal muscle weakness (patients may have difficulty getting out of a chair without assistance).
Etiology of Cushing’s syndrome
The most common cause of glucocorticoid excess is exogenous due to glucocorticoid medication. This can be due to pills, joint injections, topical creams or inhaled steroids. Non-prescription compounded medications may contain glucocorticoids. It is important to take a thorough history to assess for any steroid intake when evaluating for the cause of symptoms of cortisol excess.
Excess cortisol due to an ACTH producing pituitary adenoma is known as Cushing’s disease and is the most common cause of endogenous Cushing’s syndrome (80%). Tumors are usually <1cm (microadenoma) and may be hard to identify even by MRI. Tumors are benign and usually retain some negative feedback inhibition (ACTH is high-normal or inappropriately normal).
Excessive ACTH produced by a tumor outside the pituitary is rare (5-10% of endogenous Cushing’s syndrome). Most common are small cell tumors of the lung (malignant) and bronchial carcinoid of the lung (benign). Tumors usually do not respond to glucocorticoid negative feedback and ACTH levels (and often cortisol levels) can be quite high.
Excess cortisol can also be produced by an adrenal tumor (10-15% of endogenous Cushing’s). These tumors are usually benign, but adrenal cortical carcinomas (ACC) (malignant) may also secrete glucocorticoids. Imaging (triple phase contrast CT) can usually distinguish between the two due to differences in size (adenomas are often <4cm), shape (adenomas are round and homogeneous), and contrast wash-out (adenomas clear the contrast more quickly). ACTH levels are low with adrenal Cushing’s syndrome due to negative feedback at the pituitary from excess cortisol production.
Diagnosis of Cushing’s syndrome
Many patients have signs and symptoms that can be associated with Cushing’s syndrome, including fatigue, obesity, hypertension and diabetes mellitus. Many conditions can also cause mild increases in cortisol production, including depression, disordered eating, anxiety disorders, chronic stress or illness and alcoholism. Identifying those patients with symptoms due to pathologic cortisol excess vs. expected physiologic cortisol excess can be challenging but is important in evaluating these patients to ensure appropriate treatment.
The first step in making the diagnosis of Cushing’s syndrome is to identify cortisol excess. Normally, cortisol is secreted in a diurnal pattern, with low levels at 11pm – midnight, and highest levels first thing in the morning. With Cushing’s syndrome, levels tend to be high throughout the day. A random cortisol level is not helpful in making the diagnosis. You need to demonstrate elevated levels over a 24-hour period, or measure cortisol levels when they should be low (Figure 5).
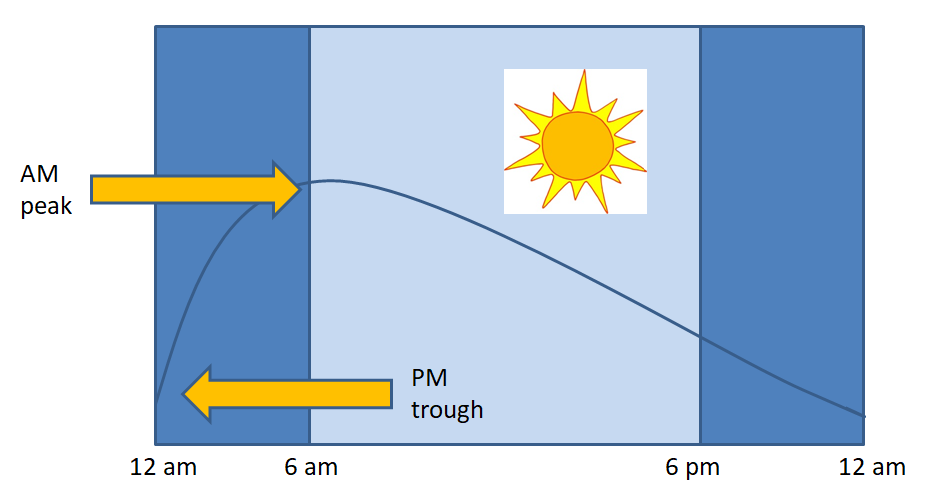
Figure 5. Normal variation in cortisol levels over a 24-hour period. Note lowest levels are between 11 pm and midnight, with highest levels between 6-8 am. If evaluating for abnormally high cortisol levels, the best time to check is when levels would normally be low (i.e. late night).
There are several screening tests used to identify hypercortisolism:
1) 24h urinary free cortisol (UFC) – Excess cortisol production overwhelms binding proteins and more free cortisol is filtered into the urine. In true Cushing’s syndrome, the urine free cortisol levels are usually 2-3 times the upper limit of normal. This test can be inaccurate in patients with renal disease.
2) Salivary cortisol – Only free cortisol is secreted into the saliva and is increased in Cushing’s syndrome due to excess cortisol production. Cortisol levels are usually low between 11pm and midnight but would be elevated in Cushing’s syndrome due to loss of diurnal variation. Normal (low) salivary cortisol between 11pm – midnight argues against Cushing’s syndrome, while an elevated value is suggestive of hypercortisolism. This test is not useful in people who do not have a normal sleep-wake cycle (shift workers, medical residents, etc.) as their cortisol pattern may be different.
3) Low dose dexamethasone suppression test – In patients with a normal HPA axis, 1 mg dexamethasone at bedtime should suppress ACTH levels, which would then suppress the normal morning cortisol level the next day to <1.8 µg/dL. Cortisol levels in patients with Cushing’s will often remain higher than expected (>5 µg/dL). The test can have a high rate of false positives due to altered metabolism of dexamethasone or cortisol (depression, stress, obesity, ethanol intake, some medications). As well, unlike the other tests which measure free cortisol, this test measures total cortisol. Thus, results could be altered in states with abnormal levels of CBG (pregnancy, oral contraceptives, exogenous estrogen, malnutrition, liver disease or acute illness).
Once elevated cortisol levels have been confirmed, the source of abnormal cortisol must be located. Plasma ACTH will help localize the source of excess cortisol production – ACTH will be low/suppressed in non-ACTH dependent Cushing’s (adrenal source) due to appropriate suppression by the high cortisol levels via the negative feedback loop. ACTH will be normal (inappropriately) or high in ACTH dependent Cushing’s (pituitary or ectopic ACTH source), as the abnormal ACTH secretion is resistant to normal suppression by cortisol.
To distinguish between pituitary and ectopic Cushing’s (both ACTH-dependent with high/inappropriately normal ACTH), several different tests can be used (see Table 3). Several of these tests use the fact that ACTH excess originating from the pituitary gland is often responsive to some feedback inhibition and stimulation (high dose steroids and CRH, respectively) whereas ACTH excess originating from an ectopic sources does not. A high dose dexamethasone suppression is one test that can be used. ACTH and cortisol are tested the morning after higher dose (8mg) of dexamethasone is taken. ACTH from a pituitary source will usually suppress with high doses of steroids, whereas ectopic ACTH-secreting tumors usually DO NOT suppress with high-dose dexamethasone. Alternatively, a CRH stimulation tests can be used. In this test, a dose of CRH (corticotrophin releasing hormone) is given which will stimulate pituitary ACTH production leading to an increase in ACTH and cortisol levels. With ectopic ACTH, cells are not responsive to CRH signaling, so no increase in ACTH or cortisol would be seen. The most sensitive and specific test (but not widely available) is a test called inferior petrosal sinus sampling (IPSS). This test involves sampling of ACTH levels from blood from the inferior petrosal sinus (which drains blood from the pituitary gland) and comparing those ACTH levels to peripheral levels of ACTH. When the IPS levels of ACTH are her (usually 3x higher) than the peripheral levels, this is consistent with cortisol excess from the pituitary gland. When the IPS levels from the IPS and periphery are equal, this is consistent with an ectopic source of ACTH. These tests can be difficult to interpret however, due to overlap between pituitary and ectopic tumors in their response to both dexamethasone and CRH. Typical results are summarized in table 3.
|
|
ACTH levels |
Low dose dexamethasone suppression test |
High dose dexamethasone suppression test |
CRH stimulation |
IPSS (interior petrosal sinus sampling) |
|
Physiologic |
Normal |
Cortisol suppresses |
n/a |
n/a |
n/a |
|
Adrenal Cushing’s |
Low |
Cortisol doesn’t suppress |
n/a |
n/a |
n/a |
|
Pituitary Cushing’s |
High (or inappropriately normal) |
Cortisol doesn’t suppress |
Cortisol suppresses |
Cortisol increases |
IPS ACTH > peripheral ACTH |
|
Ectopic Cushing’s |
Very high |
Cortisol doesn’t suppress |
Cortisol doesn’t suppress |
Cortisol doesn’t change |
IPS ACTH = peripheral ACTH |
Table 3. Results of biochemical testing for different causes of elevated cortisol levels.
Only once the biochemical workup is clear, the next step is to search for the tumor with imaging. Because both pituitary and adrenal incidental tumors are common you must complete biochemical testing prior to imaging, as the presence of a pituitary or adrenal tumor does not confirm the cause of hypercortisolism. You do not want to send a patient to surgery without confirmation of the diagnosis!
- Treatment of Cushing’s syndrome
Treatment depends on the underlying cause. With exogenous glucocorticoid use, it is important to remove the offending medication if possible. If not possible to discontinue the medication, minimize dose and duration and treat/prevent complications such as bone loss, hypertension, and diabetes.
For endogenous Cushing’s syndrome, surgery is usually the best initial option regardless of tumor location. Patients with pituitary Cushing’s who are not cured surgically may undergo radiation treatment. Rarely, these patients will undergo bilateral adrenalectomy as a last resort.
When surgery is contraindicated, there are medical options for treating Cushing’s syndrome. These medications can be divided into three different classes: corticosteroid synthesis inhibitors, adrenolytic medication, and corticosteroid receptor blockers. Corticosteroid synthesis inhibitors (ketoconazole, metyrapone, and osilodrostat) inhibit the enzymes that are involved in the synthesis of cortisol. Osilodrostat potently inhibits the 11ß-hydroxylase enzyme (responsible for the conversion of 11-deoxycortisol to cortisol). Metyrapone and ketoconazole (also an anti-fungal drug at lower doses) both act on several enzymes in the adrenal steroidogenesis pathway. These medications are often limited by the side effects of hepatotoxicity and GI side effects. When given in excess, adrenal insufficiency can occur.
Mitotane (an adrenolytic) is typically used for treatment of adrenal cortical carcinoma or cases of severe Cushing’s syndrome. Steroid hormone synthesis is stopped due to a cytotoxic effect on the adrenocortical cells leading to cell death. Mitotane also inhibits several adrenal enzymes involved in steroidogenesis. Due to its adrenolytic properties, the main side effect of mitotane is adrenal insufficiency.
Finally, mifepristone is a glucocorticoid receptor antagonist. This medication is approved in people with Cushing’s syndrome with diabetes or hypertension who are not a candidate for surgery or in whom surgery has not worked. Mifepristone blocks binding of cortisol at the receptor. Because of the receptor blocking effects, there is lack of negative feedback effect at the pituitary gland which leads ACTH to rise and there may be a compensatory cortisol response. Cortisol levels cannot be monitored to evaluate for treatment effect. Only symptoms (blood sugars, for example) can be monitored to determine clinical effect. Like the other medications discussed above, adrenal insufficiency is a known side effect.
- Adrenal insufficiency
There are two broad categories of adrenal insufficiency (AI): primary AI which is due to a problem with the adrenal glands which impairs hormone production from all layers of the adrenal cortex and secondary AI due to inadequate pituitary ACTH secretion and decreased stimulation of the adrenal glands.
- Primary adrenal insufficiency
More than 90% of both adrenal cortices need to be destroyed before symptoms of primary adrenal insufficiency occur. Initial symptoms of adrenal insufficiency can be vague, intermittent and mild (Table 4). Many patients have recurrent presentations to medical providers with dizziness, hypotension and signs of dehydration before the diagnosis is made. Importantly, the most common (postural hypotension) and specific (salt-craving) AI findings are due to insufficient aldosterone, NOT insufficient cortisol. Other signs of aldosterone deficiency include hyponatremia, hyperkalemia, and metabolic acidosis. Adrenal androgens are the primary source of androgens in pubertal children and patients without testicular androgen production, so these patients may see symptoms of low androgens (decreased or absent pubic and axillary hair, loss of libido), while patients with functioning testes, or those on exogenous testosterone therapy, would not have symptoms related to decreased DHEA production.
Patients with primary AI may also have hyperpigmentation. Cortisol deficiency leads to loss of negative feedback at the pituitary, resulting in high levels of ACTH. ACTH is synthesized from POMC. POMC is also a precursor to α-melanocyte stimulating hormone (α-MSH) which binds the MC1 receptor in skin cells and causes increased melanin production. This results in the characteristic hyperpigmentation of patients with high ACTH levels due to primary adrenal insufficiency (or ectopic ACTH production).
|
|
Symptoms |
Signs |
|
Systemic |
Weakness, fatigue, anorexia |
Weight loss, fever, hyperkalemia, metabolic acidosis, hyponatremia |
|
Skin |
|
Hyperpigmentation |
|
GI |
Nausea, abdominal pain, salt-craving |
|
|
CVS |
Palpitations, dizziness |
Tachycardia, hypotension (postural) |
Table 4. Signs and Symptoms of Primary AI
Although patients can often tolerate adrenal insufficiency for a long time, an infection, injury, or major stress can cause rapid clinical deterioration with hypotension and death if this “adrenal crisis” is not diagnosed and treated in time.
The incidence of primary adrenal insufficiency is about 1-2 per million patients. Autoimmune AI (Addison’s disease) is the most common cause of primary adrenal insufficiency in the US, accounting for about 75% of cases. A lymphocytic adrenalitis destroys the cortex which becomes fibrotic and thin. Autoantibodies (eg. anti 21-hydroxylase) are found in 50-70% of patients. About 50% of patients with autoimmune adrenal insufficiency have, or develop, one or more other autoimmune disorder, such as lymphocytic thyroiditis, vitiligo, type 1 diabetes, or pernicious anemia.
Adrenal glands have a predisposition for mycobacterial infections and mycobacterium tuberculosis is the most common cause of primary adrenal insufficiency world-wide. The infection usually destroys both adrenal glands and can form bulky granulomatous infiltrates. Immunosuppression is a risk factor for mycobacterial adrenalitis. In patients with AIDS, primary adrenal insufficiency is most commonly due to mycobacterium avium, but many other infections including primary HIV, fungal, atypical bacterial and viral infections have been associated with adrenal insufficiency. Always consider adrenal insufficiency in patients with AIDS admitted with hypotension, hypoglycemia, fever or wasting.
Several infiltrative processes can destroy the adrenal glands, such as amyloidosis (deposition of abnormal proteins) or hemochromatosis (the deposit of iron). Bilateral adrenal hemorrhage can cause adrenal failure, most diagnosed in older patients receiving chronic anticoagulation. Adrenal hemorrhage is also seen as a complication of Neisseria meningitis, known as Waterhouse-Friderichsen syndrome (WFS). These patients have adrenal failure due to bilateral adrenal hemorrhage related to the underlying bacterial infection. The adrenal gland is also a common site of metastatic disease from primary cancers of the skin, lung, breast, or stomach. Metastatic adrenal disease is often discovered incidentally on abdominal CT scan as these adrenal masses are usually asymptomatic and even when large and bulky only rarely cause enough adrenal destruction to result in adrenal insufficiency.
Secondary adrenal insufficiency
Symptoms of secondary adrenal insufficiency are similar to primary adrenal insufficiency with a few exceptions. Orthostasis and salt craving are usually not present in secondary AI, because aldosterone secretion is not dependent on ACTH and mineralocorticoid levels are maintained in secondary AI. Hyperpigmentation is not present, because ACTH (and thus αMSH) levels are low not elevated.
The most common overall cause of adrenal insufficiency in highly developed countries is discontinuation of exogenously administered glucocorticoid medications. Chronic use of glucocorticoids suppresses ACTH production which leads to secondary adrenal insufficiency. Chronic suppression of ACTH leads to atrophy of the adrenal glands. If these medications are abruptly discontinued, it can take some time for the adrenal glands to recover normal function, and patients may have transient secondary AI.
Hypothalamic and pituitary disorders, which disrupt normal ACTH production, are another cause of secondary AI. Pituitary tumors and non-pituitary tumors near the sella turcica, as well as surgical and radiation treatment of these tumors can cause injury to the HPA axis and cause secondary adrenal insufficiency. Though pituitary adenomas are common, it is rare for them to cause secondary adrenal insufficiency. The HPA axis is usually the most resistant of the pituitary hormonal axes to pressure from a tumor.
Traumatic brain injury, pituitary hemorrhage or apoplexy (infarction), or Sheehan’s syndrome due to peri-partum pituitary ischemia, can cause secondary AI, as can infiltrative and autoimmune diseases of the pituitary and pituitary stalk (i.e. sarcoid, histiocytosis (various types), lymphocytic hypophysitis, neurosyphilis).
- Diagnosis of adrenal insufficiency
Because of the variability in serum cortisol throughout the day, a random cortisol level will seldom be helpful to assess adrenal function. Adrenal insufficiency may be suspected if morning cortisol levels are low (as they should typically be high at this time of day) (figure 5). However, using dynamic testing such as Cosyntropin stimulation test to stimulate the adrenal gland is required to confirm the diagnosis.
With AI, either primary due to adrenal damage or secondary due to chronically low ACTH, the adrenal gland is unable to respond to normal stimulation by ACTH. In the case of secondary AI, this is due to atrophy of the adrenal gland due to lack of stimulation by ACTH. To confirm AI, baseline cortisol levels are measured and the patient is given a dose of synthetic ACTH (Cosyntropin). After 60 minutes, the normal adrenal gland will respond with a significant increase in cortisol levels. However, with AI the cortisol levels will remain low. There is one caveat to this. In the immediate (0 to ~ 14 days) period after pituitary or hypothalamic damage and subsequent ACTH deficiency, the adrenal glands have not yet atrophied to a significant degree. In this case the Cosyntropin response will be falsely ADEQUATE even when complete secondary adrenal insufficiency is present. This is most likely to occur with pituitary apoplexy, when pituitary damage occurs rapidly.
Once the diagnosis of AI is confirmed, ACTH levels are used to distinguish between primary causes (high ACTH) and secondary causes (low ACTH). This distinction is important, as patients with primary AI are usually deficient in both mineralocorticoids and glucocorticoids due to damage to the adrenal cortex, while patients with secondary AI only need glucocorticoid replacement.
Treatment of adrenal insufficiency
The primary goal for treatment of adrenal insufficiency is to provide adequate hormone replacement to prevent symptoms of adrenal insufficiency while minimizing side effects. All patients with adrenal insufficiency, regardless of cause, need glucocorticoid replacement which is typically done with hydrocortisone or prednisone. Less often, dexamethasone is used. Often we try to mimic the normal secretion of cortisol by giving hydrocortisone twice daily (with a higher morning dose to mimic diurnal variation), but some patients prefer to take prednisone once a day for convenience. Symptoms are a more useful indicator of success than following ACTH or cortisol levels. Cortisol replacement needs to be increased during acute stress either at home (i.e. fever/infection) or on admission to hospital (surgery, trauma, acute illness, etc.) to mimic the normal stress response (physiologic cortisol increase with stress).
Patients with primary AI also need mineralocorticoid replacement. Most patients can be treated with 0.1 mg of fludrocortisone daily. Fludrocortisone acts at the mineralocorticoid receptor with only minimal glucocorticoid activity and replaces aldosterone in patients who have primary AI. Patients with chronic secondary adrenal insufficiency rarely need fludrocortisone because mineralocorticoid secretion remains intact due to normal angiotensin signaling.
Replacement of adrenal androgens is not necessary. There is no strong evidence that replacement of DHEA has any impact on patient well-being.
- Congenital Adrenal Hyperplasia (CAH)
Another adrenal disorder that can result in low cortisol levels is congenital adrenal hyperplasia. In CAH, blockade of cortisol synthesis (Figure 2) is due to gene mutations encoding key enzymatic steps. These genetic disorders are inherited in an autosomal recessive fashion. Decreased cortisol synthesis results in increased ACTH secretion (due to loss of negative feedback), which leads to hyperplasia of the adrenal cortex (due to the trophic effect of ACTH) and increased synthesis of adrenal steroids upstream of the enzymatic blockade. The phenotypic variability of CAH depends on the mutated gene and the degree to which the mutation impairs gene function. Some individuals present with severe cortisol and aldosterone deficiency at birth (salt-wasting CAH) while others have no evidence of hormone deficiency in childhood (non-classical CAH) and the diagnosis is made at the time of puberty due to androgen excess (amenorrhea, acne, hirsutism). This is covered in detail later in the Foundations phase when you study sex hormones. The long-term management of CAH involves treatment of adrenal insufficiency and/or suppression of pituitary ACTH with exogenous glucocorticoids to avoid over production of unaffected hormones, while avoiding the negative side effects that come with too much glucocorticoid treatment.
- Pathology of the adrenal medulla
- Tumors of the adrenal medulla
Pheochromocytomas are tumors of the adrenal medulla that make, store, and secrete excessive amounts of catecholamines. When they occur in extra-adrenal chromaffin tissue they are known as paragangliomas. Adrenal pheochromocytomas usually have both epinephrine- and norepinephrine-containing granules while extra-adrenal paragangliomas usually contain only norepinephrine because these tumors are not exposed to high levels of cortisol (recall that PNMT is induced by glucocorticoids), and thus PNMT is unable to convert norepinephrine to epinephrine. Most pheochromocytomas (90%) are benign.
- Neuroblastomas are another tumor of the adrenal medulla and are the most common abdominal tumor in infancy and childhood. Many neuroblastomas secrete catecholamines and catecholamine metabolites. About one in five patients with neuroblastoma is hypertensive from excessive catecholamine secretion. Ganglioneuroblastomas and ganglioneuromas are solid tumors that may occur in sympathetic ganglia or the adrenal medulla, and they may synthesize catecholamines. These tumors should be treated as if they were pheochromocytomas.
Clinical features of pheochromocytoma
The clinical features of pheochromocytomas are due mainly to the release of catecholamines. Hypertension is the most common sign (90% of patients), but pheochromocytoma is a rare cause of hypertension, occurring in only 0.1% of the hypertensive population. Hypertension may be sustained (60%) or paroxysmal (40%) and is mediated largely through the interaction of norepinephrine and its receptors. Other symptoms or clinical signs observed with pheochromocytoma are also explained by the hyper-stimulation of adrenergic receptors in a variety of tissues and organs. These manifestations include palpitations, sweating, headache, nervousness, tremulousness, hyperglycemia, chest pain or abdominal pain, and nausea. About 25% of pheochromocytoma cases are hereditary, seen with von Hippel Lindau syndrome, neurofibromatosis, MEN 2A/ 2B and with mutations in the succinyl dehydrogenase genes: SDHB, SDHC and SDHD.
Diagnosis of pheochromocytoma
Since catecholamine secretion can be increased by several physiological stimuli, establishing abnormal elevation of plasma catecholamines can be difficult. Plasma tests for the diagnosis of pheochromocytoma historically have been fraught with several problems. Venipuncture may stimulate a surge in catecholamine secretion leading to elevations of plasma catecholamines even in patients without a pheochromocytoma. Caffeinated beverages and some medications (e.g., tricyclic antidepressants) may also lead to false-positive elevations in plasma catecholamine tests. On the other hand, serum catecholamine levels might be normal in patients with a pheochromocytoma that secretes only very small amounts or only intermittently secretes excess catecholamines.
The measurement of plasma free metanephrines has been shown to be useful in the diagnosis of pheochromocytoma. Virtually all pheochromocytomas continuously secrete excessive metanephrines (reflecting internal metabolism of catecholamines in the tumor). Therefore, a normal plasma free metanephrine test makes a pheochromocytoma extremely unlikely. An alternative to blood tests would be a 24-hour collection for urine catecholamines and metabolites. This is a more sensitive and specific test for diagnosis of pheochromocytoma and is typically used to confirm the diagnosis when plasma tests are positive. Clinical and biochemical diagnosis of pheochromocytoma should always be done before attempts to radiographically localize the tumor, with CT scan or MRI, due to the high incidence of benign, non-functioning adrenal nodules, especially in older adults.
- Treatment of Pheochromocytoma
Definitive treatment of a pheochromocytoma is surgical excision of the tumor. In preparation for surgery, patients are usually treated with α1-adrenergic receptor antagonist (doxazosin or phenoxybenzamine) and ß-adrenoreceptor blockers. It is important to start alpha blockade prior to starting beta blockade. Blocking the vasodilating effects of the beta-2 receptors prior to blocking the vasoconstricting alpha-1 receptors can precipitate a hypertensive crisis. ß-adrenoreceptor blockers may be needed (but not always) to treat the reflex tachycardia that is associated with alpha-1 blockade.
Feedback/Errata